Markery stresu
i zaburzeń metabolizmu komórkowego
Stres oksydacyjny
Stres oksydacyjny to zaburzenie homeostazy, prowadzące do zaburzenia równowagi prooksydacyjno – antyoksydacyjnej w kierunku reakcji utleniania. W wyniku tego dochodzi do dezintegracji i uszkodzenie komórek, co skutkuje zaburzeniem ich funkcjonowania.
Nasza oferta zawiera pakiet 3 badań diagnozujących stres oksydacyjny:
Istnieją teorię, że stres oksydacyjny może odgrywać ogromną rolę w powstawaniu zaburzeń autystycznych. Antyoksydanty mogą poprawiać obraz choroby u tych pacjentów. Ważna w takim przypadku jest suplementacja substancjami ochronnymi, do których należą: witamina C i E, beta-karotyna, koenzym Q10, selen, cynk, miedź, mangan, substancję roślinne z grup karotynidów, polifenoli, fitoestrogenów i sulfidów. Działają one jako wychwytywacze wolnych rodników, chelatują metale i chronią komórki przed utlenieniem.
Równie poważnym zaburzeniem co stres oksydacyjny jest stres azotowy polegający na nadmiernym tworzeniu tlenku azotu (NO) i produktów jego metabolizmu: peroksynitrytu, nitrotyrozyny i kwasu nitrofenylooctowego.
Tlenek azotu (NO) jest wolnym rodnikiem, który jest tworzony niemal we wszystkich komórkach ludzkiego organizmu. Z powodu swoich niewielkich rozmiarów i wysokiej lipofilności przenika on szybko przez bariery. Ma on krótki czas działania ale wysoką aktywność biologiczną. Z tlenem reaguje szybko przechodząc w nitraty.
Tworzenie tlenku azotu (NO) zachodzi poprzez enzym – syntetazę tlenkoazotową (NOS) z L-argininy. Jako produkt uboczny tej rekcji powstaje cytrulina i woda.
Tlenek azotu pełni ważne funkcje fizjologiczne. Syntetaza tlenkowęglowa występuje w czterech postaciach izomerycznych:
Indukowalna NOS (iNOS)
– znajduje się w komórkach systemu immunologicznego (neutrofile, makrofagi). Powstający dzięki niej tlenek azotu działa cytotoksycznie na bakterie. Działanie iNOS jest indukowane poprzez cytokiny a także poprzez różne leki oraz obce substancje chemiczne.
Śródbłonkowa NOS (eNOS)
– znajduje się w śródbłonku naczyń krwionośnych. Powstający NO działa naczyniorozszerzająco oraz oskrzelorozszerzająco poprzez aktywację guanylocyklazy i wpływ na tworzenie cGMP.
Neuronalna NOS (nNOS)
class="mb-3" – tworzy NO w komórkach nerwowych. Indukuje to powstawanie glutationu w synapsach i dzięki temu uczestniczy w neurotransmisji bodźców.
Mitochondrialna NOS
– reguluje metabolizm mitochondriów niezbędny dla tworzenia komórek, proliferacji, apoptozy oraz oddychanie komórkowe. Z powodu wysokiego powinowactwa do grup hydroksylowych, peroksylowych, tyrozynowych NO jest wolnym rodnikiem. W fizjologicznych stężeniach hamuje on oksydację lipidów. Kolejnymi zadaniami fizjologicznym są hamowanie agregacji trombocytów i wzmaganie odpowiedzi na insulinę.
Wpływ na energetyczny bilans komórkowy
Cykl Krebsa stanowi centralny punkt przemiany materii. W nim odbywa się rozkład węglowodanów tłuszczy i białek do Ac-CoA. Substancja ta jest metabolizowana w celu pozyskania energii. Powstaje przy tym GTP oraz produkty reakcji: NADH i FADH , które są dalej przetwarzane w ATP. Cykl Krebsa jest także poprzez swoje produkty pośrednie punktem wyjścia do rozmaitych biosyntez. Temu służy np. Alfa-ketoglutaran jako substancja wyjściowa do tworzenia aminokwasów, glutaminy, argininy, proniny. Z bursztynylo-CoA jest wytwarzany kwas deltaaminolewuliunowy, który dalej jest substancją bazową do biosyntezy hemu.
Wysokie poziomy NO hamują enzym akonidazę i uniemożliwiają przemianę cytrynianu i izocytrynianu. Powoduje to blokadę całego cyklu Krebsa i uniemożliwia metabolizowanie białek , tłuszczy i aminokwasów. W tym mechanizmie dochodzi do niedoboru NADH i FADH2 i wynikający stąd niedobór substratów do syntezy ATP w mitochondriach. W konsekwencji w komórkach pojawia się deficyt energetyczny.
Dodatkowe przyspieszenia przemiany materii np. poprzez fizyczny, psychiczny stres, infekcje lub odżywanie bogate w węglowodany wzmagają wielokrotnie syntezę NO. Powstaje ekstremalnie wysoki deficyt energii w którym dochodzi także do blokady oksydacji kwasów tłuszczowych oraz blokady rozkładu węglowodanów (glikolizy). W procesie glikolizy z glukozy tworzony jest kwas pirogronowy oprócz tego jest on produktem ubocznym rozkładu aminokwasów: glicyny, cysteiny, alaniny. Przy wystarczającym poziomie energii w komórce, przy pomocy enzymu dehydrogenazy pirogronowej dochodzi do jego przemiany w Ac-CoA. W sytuacji stresu azotowego ten ważny szlak metaboliczny jest uszkodzony z powodu niedostatecznej aktywności enzymatycznej. Prowadzi to do osłabienia wydajności energetycznej komórek. Kwas pirogronowy zamiast przemiany w Ac-CoA jest redukowany do kwasu mlekowego. Mając na uwadze to zjawisko można mierzyć zaburzenia metabolizmu poprzez określanie stosunku kwasów: mlekowego do pirogronowego. Im wyższe wartości przyjmuje ten wskaźnik tym bardziej zaburzony jest bilans energetyczny.
Inne działania tlenku azotu (NO)
Jakiekolwiek komórki produkujące cytokiny mogą stymulować NO. Nie wiadomo, czy nadmierna produkcja NO w autyzmie jest umiejscowiona w konkretnych organach czy tkankach. Najprawdopodobniej ma to miejsce w mózgu i przewodzie pokarmowym, oba te układy w autyzmie funkcjonują nieprawidłowo, w obrazie pojawiają się objawy behawioralne i gastrologiczne.
Nadmierny poziom NO w mózgu zwiększa apoptozę, uszkadza barierę krew-mózg, zwiększa neurodegenerację i demielinizację . Takie mechanizmy mogą mieć wpływ na obraz choroby w zespole autystycznym.
Zwiększenie produkcji NO zachodzi także poprzez ekspozycję na obce substancje: chemikalia, metale ciężkie, leki (antybiotyki, cytostatyki, statyny). Wielu autorów doszukuje się związku pomiędzy ekspozycją na metale ciężkie (szczególnie rtęć) oraz spożywaniem acetaminofenu (paracetamol) zarówno przez dzieci jak i przez matki w ciąży. Substancje te wywołują w organizmie stres azotowy.
Ekscytotoksyczność
Nadmierne tworzenie NO zaburza metabolizm. NO posiada silne powinowactwo do żelaza i siarczku żelaza co hamuje działanie enzymów. Duże ilości NO hamują cykl oddechowy mitochondriów. Spowodowana tym utrata ATP dotyka w szczególności komórki o wysokim zapotrzebowaniu energetycznym a więc komórki nerwowe i komórki układu immunologicznego. W rezultacie aktywują się receptory glutaminianowe w tkance nerwowej (synapsach). Glutaminian wypiera blokujący kanał jon magnezu i otwiera go powodując napływ jonów wapnia do komórki nerwowej. W stresie azotowym proces ten jest silnie wzbudzony. Powoduje to utratę funkcji komórki nerwowej i częściową blokadę przewodnictwa. Dłużej trwający napływ wapnia prowadzi do obumierania komórek. Zjawisko takie nazywa się ekscytotoksycznością.
Nadmiar wapnia uruchamia aktywację syntazy tlenku azotu (iNOS) i kinazę białkową C (PKC). iNOS powoduje produkowanie w nadmiarze tlenku azotu, który zaczyna kumulować się w komórkach. Gdy napotka wolne rodniki tworzy bardzo destrukcyjną cząsteczkę, która uszkadza mitochondria – główne źródło energii dla neuronu.
W tym samym czasie kinaza białkowa C aktywuje fosfolipazę A2 w membranie komórkowej, co sprawia że kwas arachidonowy jest wypuszczany do cytozolu. Tam działają na niego dwa enzymy: lipoksygenaza i cyklooksygenaza, które produkują potencjalnie destrukcyjne eikozanoidy. Najistotniejszym jest enzym COX II, który prowadzi do kumulacji prostaglandyn PGE2 i PGD2, które wzmagają stan zapalny.
Kumulacja eikozanoidów prowadzi do produkowania wolnych rodników, w szczególności bardzo destrukcyjne rodniki hydroksylowe. Proces ten przyspiesza i wolne rodniki wchodzą w interakcje z membranami neuronów, w tym membranami mitochondrialnymi i komórkowymi. Gdy ten proces się zacznie, początkuje on reakcję łańcuchową w wielonasyconych kwasach tłuszczowych, z których zrobione są membrany. Proces ten nazywamy peroksydacją lipidową i jest to objaw stresu oksydacyjnego. Jak widać zatem: obydwa rodzaje stresu: tlenowy i azotowy są ze sobą powiązane.
Aktywacja układu immunologicznego
Aktywacja mikrogleju i astrocytów w procesie zapalnym, poprzez stymulację układu odpornościowego może wywołać ekscytotoksyczność. Aktywacja mikrogleju powoduje uwolnienie się licznych cytokin, w tym TNF-alfa, IL-1ß, IL-2, IL-6 i INF-gamma. Dodatkowo, ma miejsce aktywacja prozapalnych eikozanoidów. Powiązane z tym procesem jest wygenerowanie różnego rodzaju produktów pośrednich przemian tlenowych i azotowych, w tym nadtlenku wodoru, rodników wodorotlenowych, nadtlenoazotynu i 4-hydroksynonenalu. Te produkty pośrednie nie tylko uszkadzają neurony, połączenia synaptyczne i części składowe komórek ale indukują też uwolnienie glutaminianu z sąsiadujących astrocytów.
Aktywacja mikrogleju może również spowodować uwolnienie się glutaminianu i kwasu chinolinowego – dwóch silnych ekscytotoksyn – z samego makrofagu. Jest to związane z uruchomieniem alternatywnego szlaku metabolizmu tryptofanu. Wszystko, co prowadzi do aktywacji mikrożelu, w tym wirusy, ß-amyloidy, rtęć, aluminium, oksydowany LDL i HDL, homocysteina i ekscytotoksyny, może wpłynąć na kumulację kwasu chinolinowego. Szczególnie istotna jest równowaga między kwasem chinolinowym a kynurenitami, które pełnią dla mózgu funkcję ochronną.
Interakcje z bakteriami, wirusami i lipopolisacharydami mogą zwiększyć wydzielanie się glutaminianu 2-3 krotnie powyżej normalnego poziomu. Należy również zauważyć, że nadmiar glutaminianu, tak jak i jego niedobór, wpływa na długoterminowe wzmocnienie kluczowe dla procesów uczenia się i zapamiętywania. Dodatkowo, wzrost i kierunek rozwoju ścieżek w mózgu jest również regulowany przez glutaminian, w szczególności wpływ na niego ma przedłużający się okres nadmiaru glutaminianu. Niedobór glutaminianu zaburza funkcję stożka wzrostu i prowadzi do niewłaściwej budowy mózgu.
Oksydacja lipidów
Podczas oksydacji lipidowej powstają produkty uboczne w tym kilkanaście różnych aldehydów. Najbardziej powszechny jest malondialdehyd (MDA), a najbardziej destrukcyjny jest 4-hydroksynonenal (4-HNE). Ostatnie badania dowiodły, że 4-HNE może w dużym stopniu niszczyć komórki, włącznie z zahamowaniem defosforylacji białka tau, co wpływa na funkcję mikrotubuli. Dowiedziono także, że hamuje on reduktazę glutationową, która jest niezbędna do przemiany utlenionego glutaminianu do jego funkcjonalnej, zredukowanej formy. Stwierdzono, że dzieci z chorobami autoimmunologicznymi mają wyższe poziomy 4-HNE we krwi niż dzieci z grupy kontrolnej.
Stwierdzono również, że 4-HNE może łączyć się z proteinami synaps, gdzie upośledza transport zarówno glukozy, jak i glutaminianu. Ten proces jest wyjątkowo niebezpieczny, bo liczne badania wykazały, że upośledzenie źródeł energii znacznie zwiększa podatność na działanie glutaminianu do tego stopnia, że nawet normalne poziomy glutaminianu mogą być ekscytotoksyczne.
Zjawiska degeneracyjne
Wiadomo już również z badań doświadczalnych, że napady drgawkowe są blisko związane z procesem ekscytotoksyczności . Nie tylko glutaminian i kwas aspartamowy mogą wywoływać drgawki, ale same drgawki mogą stymulować uwalnianie się ekscytotoksyn w mózgu, prawdopodobnie przez stymulację powstawania wolnych rodników. Spontanicznie niszczone neurony, w szczególności gdy proces ma miejsce przez długi czas, powodują utratę energii, niedokrwienie i niedotlenienie, które to powodują również nadmierne uwalnianie się glutaminianu.
Podczas gdy degeneracja neuronów może brać się z zbyt wysokich poziomów glutaminianu, utrata połączeń dendrytowych może mieć miejsca przy dużo mniejszych stężeniach. Wiadomo również, że niedotlenienie i niedokrwienie, częste przy drgawkach występujących w długim okresie czasu, mogą również drastycznie zwiększać poziom glutaminianu. Może mieć to duże znaczenie przy formowaniu się ścieżek, jak również wpływa na utratę neuronów, połączeń synaptycznych i komórek pnia mózgu. Wiadomo, że po osiągnięciu wieku 2 lat rozwijający się mózg zawiera więcej receptorów glutaminianu niż po urodzeniu i ta liczba powoli zmniejsza się przez następną dekadę. Dlatego mózgi dzieci są bardziej podatne na ekscytotoksyczność.
Cytrulina, kwas metylomalonowy, kwas nitro fenylooctowy
Cytrulina
– jest produktem ubocznym cyklu mocznikowego, z którego z tlenu i argininy powstaje tlenek azotu. Jest więc markerem stresu azotowego, w chwili nadmiernej produkcji NO.
Kwas metylomalonowy (MMA)
– jest wytwarzany w trakcie metabolizmu aminokwasów w niewielkich ilościach. Jest on bardzo dobrym i czułym wskaźnikiem niedoborów witaminy B12, której zadaniem jest przekształcanie metylomalonylo-CoA w bursztynylo-CoA. W chwili, gdy w organizmie jest niewystarczająca ilość witaminy B12 nadmierna ilość metylomalonylo-CoA jest przekształcana do kwasu metylomalonowego. Zwiększona ilość MMA może być przyczyna kwasicy metylomalonowej, która jest skutkiem wrodzonej wady metabolicznej, nie daje ona objawów bezpośrednio po urodzeniu jednak wraz ze spożyciem większej ilości białek i powstawania nadmiernej ilości MMA mogą wystąpić m.in. napady padaczkowe, zaburzenia rozwoju.
Kwas nitrofenylooctowy
class="mb-3" – jest produktem metabolizmu tlenku azotu. Powstaje w wyniku przyłączenia NO do aromatycznych aminokwasów, w wyniku czego powstaję nitrotyrozyna, która przekształca się do kwasu nitrofenylooctowego. Powinowactwo NO do tyrozyny i tryptofanu ma szczególnie negatywny wpływ na produkcję hormonów i neurotransmiterów. Może być wykorzystywany także jako marker stresu azotowego.
Nitrotyrozyna
W stresie azotowym dochodzi do tworzenia się peroksynitrytu (ONOOˉ) – wskutek nadmiaru NO lub niedostatecznej aktywności manganozależnego enzymu dysmutazy nadtlenku. Pozostający w nadmiarze ONOOˉ ma duże powinowactwo do aminokwasów szczególnie do tyrozyny i tryptofanu. W połączeniu z tyrozyną tworzona jest nitrotyrozyna, która jest związkiem bardzo stabilnym i nieulegającym dalszemu metabolizmowi. Jest to zatem dobry wskaźnik stresu azotowego.
Badamy nitrotyrozynę metodą wysokosprawnej chromatografii cieczowej, która jest złotym standardem w oznaczaniu tego związku, daleko przewyższającym metody immunoenzymatyczne.
Kwas mlekowy/kwas pirogronowy
Kwas pirogronowy jest jednym z podstawowych substratów w cyklu Krebsa w tworzeniu Ac CoA. Jego zmniejszony poziom świadczy o niedostatecznym trawieniu węglowodanów, zwiększenie poziomu świadczy o niedostatecznej funkcji enzymu przekształcającego do Ac CoA – co ma bezpośredni wpływ na wydajność energetyczną komórek. W sytuacji tego bloku zamiast do Ac-CoA kwas pirogronowy redukowany jest do mleczanu. Wzrost wartości ilorazu kwas mlekowy/kwas pirogronowy jest markerem niewydolności energetycznej organizmu poprzez blokadę cyklu Krebsa. Oba parametry oznaczane są metoda spektrometrii masowej zapewniającej najwyższą możliwą dokładność pomiaru.
Kompleksowy test neuroorganiczny – Organix-neuro
Schorzenie zapalne układu nerwowego. Znaczenie stosunku kinureniny-tryptofanu
Wiele danych klinicznych i biochemicznych dowodzi, że podstawowy aminokwas - Tryptofan stanowi „Missing link” między schorzeniami somatycznymi i psychicznymi.
Tryptofan stanowi podstawowy aminokwas, metabolizowany w warunkach fizjologicznych w serotoninę i melatoninę. We krwi tryptofan wiąże się w 80 do 90 % z albuminą i w osoczu występuje jedynie niewielka jego ilość. Jedynie około 1 procent przyjętego tryptofanu jest przekształcany w serotoninę. Przy tym największy udział syntezy serotoniny ma miejsce w komórkach enterochromafinopodobnych przewodu pokarmowego, a tylko bardzo nieznaczna część w centralnym układzie nerwowym.
Krążący w organizmie tryptofan konkuruje o wchłonięcie do mózgu z innymi aminokwasami. Dostępność tryptofanu dla neuronów serotoninergicznych jest zależna od stosunku pomiędzy tryptofanem, a stężeniami tych konkurujących aminokwasów we krwi. Wzmożoną synteza serotoniny można zatem osiągnąć zarówno podnosząc poziom wolnego tryptofanu jak poprzez obniżenie poziomu aminokwasów konkurujących z tryptofanem o przeniknięcie do mózgu (np. spadek waliny, leucyny i izoleucyny po posiłku w wyniku zwiększonego dopływu węglowodanów – spowodowany przez stymulację wydzielania insuliny oraz od-insulinowe zwiększone wchłanianie przede wszystkim waliny, leucyny i izoleucyny przez mięśnie, por. Hüther et al., 2000).
W warunkach fizjologicznych prawie jeden procent tryptofanu zostaje przetransferowany przez enzym tryptofan-5-hydroksylazę (TPH) do 5-hydroksytryptofan (5-http). Ta ścieżka metabolizmu potrzebuje jako kofaktorów kwasu foliowego, witaminy B3, żelaza, miedzi i witaminy C. Hiperinsulinemia, hiperkortyzolizm (stres) i niedobór witamin B3 oraz B6 prowadzą do blokady enzymów. Powstający w ramach stresu azotowego azotyn nadtlenowy również blokuje hydroksylazę tryptofanu. 5-http jest następnie transferowany w serotoninę przez aromatyczną karboksylazę aminokwasu L. Witamina B6 działa przy tym jako kofaktor.
Rozpad tryptofanu ma miejsce w sposób fizjologiczny dzięki wątrobowej 2,3-dioksygenazie tryptofanowej (TDO) za pośrednictwem ścieżki metabolizmu kinureniny (patrz ilustracja 2). 2,3-dioksygenaza tryptofanowa jest indukowana przez tryptofan, kinureninę i hormony steroidowe jak kortyzol i estrogeny, zaś hamowana przez NADPH.
Wpływ na energetyczny bilans komórkowy
Kwas 5-hydroksyindolooctowy (5-HIAA) stanowi główny metabolit serotoniny. Podwyższone stężenia występują w przypadku karcynoidów (guzów neuroendokrynnych) lub w leczeniu za pomocą inhibitorów wychwytu zwrotnego serotoniny (SSRI – Selective Serotonin Reuptake Inhibitor).
Ilustracja 1 ukazuje, że do początkowej hydroksylacji tryptofanu jest potrzebna jako kofaktor tetrahydrobiopteryna (BH4). Zredukowane poziomy BH4 mogą być uwarunkowane przez mutacje genetyczne w enzymach uczestniczących w syntezie-regeneracji BH4, a to ze względu na niedobór kwasu foliowego, zwiększonego stresu oksydacyjnego oraz podczas metabolicznych stanów zapalnych.
Pacjenci z obniżonym statusem BH4 wykazują progresywne opóźnienie psychomotoryczne, dystonię oraz ciężki niedobór dopaminy i serotoniny z obniżonym poziomem 5-HIAA oraz kwasu homowanilinowego (Bonafe et al., 2001). Homowanilina stanowi najważniejszy metabolit dopaminy.
TryptofanFe, wit. B3, BH 4, hydroksylaza tryptofanowa (TPH)
⇩
5-hydroksy-tryptofan
wit. B6, aromatyczna karboksylaza aminokwasu L.
⇩
Serotonina ⇨ kwas 5-hydroksyindolooctowy
⇩
N-acetyloserotonina
⇩
Melatonina
(podwyższony poziom5-hydroksy-tryptofanu)
- Leczenie za pomocą inhibitorów wychwytu zwrotnego serotoniny (SSRI)
- Nowotwory komórek enterochromafinopo-dobnych (jelito cienkie, wyrostek robaczko-wy, jelito grube, odbyt)
- Pożywienie zawierające serotoninę (np. pomidory, banany, kakao)
- Leki (rezerpina, metamfetamina)
- Ksenobiotyki jak akrylamidy (Egorava et al., 1998)
(obniżony poziom5-hydroksy-tryptofanu)
- Niewydolność nerek
- Alkohol
- Niskie poziomy BH4
- Obstrukcja ze zredukowanym obrotem serotoniny
Ilustr. 1: metabolizm tryptofanu-serotoniny
Kwas ksanturenowy
Wytwarzanie kwasu ksanturenowego ma miejsce w trakcie metabolizmu tryptofanu za pomocą metabolizmu kinureniny. Niedobór witaminy B6 prowadzi do spowolnienia ściśle zależnego od witaminy B6 postępu syntezy ze skutkiem akumulacji hydroksykinureniny i kinureniny (Chiang et al., 2005; Tada et al., 1968; Kosters et al., 1976). Oba te metabolity są szybko metabolizowane dalej w kwas ksanturenowy oraz kwas kinureninowy i pojawiają się w podwyższonych ilościach w moczu (patrz ilustr. 2; Knapp, 1961; Abbassy 1959). Podwyższone poziomy kwasu ksanturenowego świadczą o niedoborze witaminy B6, zwłaszcza wtedy, gdy wydalanie kwasu chinolinowego w moczu nie jest podwyższone. Kwas kinureninowy jest wydalany w niewielkich ilościach, ponieważ jest ona dalej metabolizowany w cyklu kwasu cytrynowego (Matte et al., 2001).
Podwyższone poziomy kwasu ksanturenowego mogą stanowić skutek niedoboru witaminy B6, modulacji metabolizmu kinureniny przez hormony steroidowe oraz skutek zredukowanego wskaźnika rotacji tryptofanu w wyniku zmniejszonego dostarczania protein.
Podwyższone poziomy kwasu ksanturenowego indukują wolne rodniki. Hydrolizowaną strukturę choinową kwasu ksanturenowego wiąże żelazo i tworzy w ten sposób kompleks uszkadzający oksydacyjnie DNA (Murakami et al. 2006).
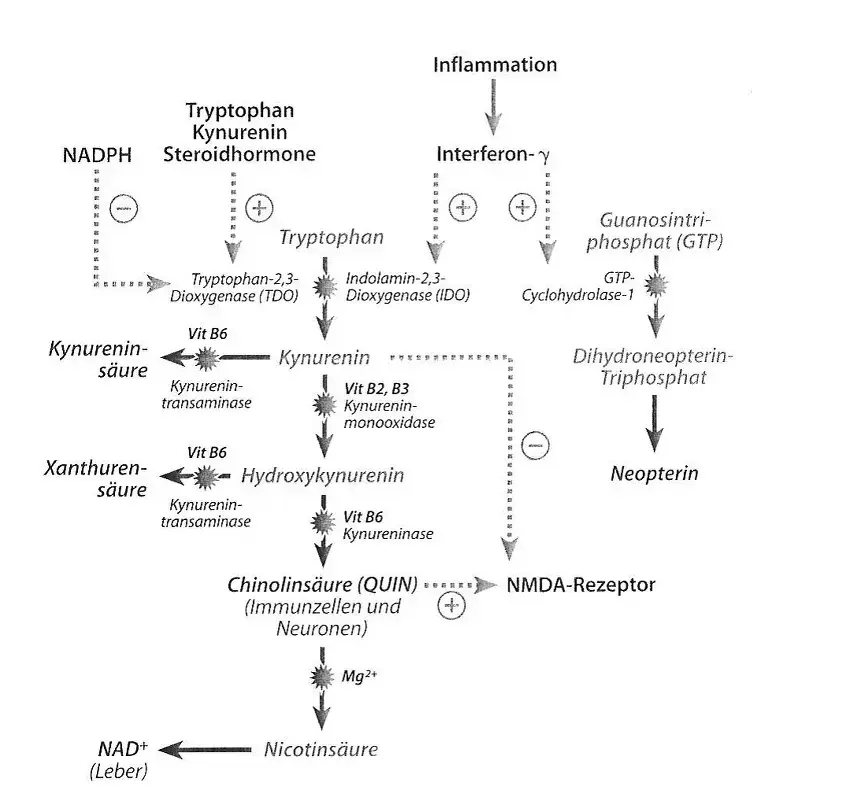
Ilustr. 2: Rozpad tryptofanu ma miejsce w warunkach fizjologicznych oksydacyjnie dzięki 2,3-dioksygenazie tryptofanowej (TDO). W zależności od tkanki powstaje przy tym produkt końcowy NAD+ (wątroba) lub kwas chinolinowy (komórki odpornościowe i neurony). W okolicznościach patologicznych (chorobowych) jak np. stan zapalny, zostaje zaindukowana 2,3-dioksygenaza indolaminowa (IDO), co prowadzi do wzmożonego wytwarzania kwasu chinolinowego. Jeśli aktywność enzymów w wyniku braku kofaktorów (witamina B2, B3, B6) zostanie obniżona lub też TDO zostanie wzmocnione w swej aktywności przez hormony steroidowe lub wzmożony metabolizm tryptofanu, to dochodzi do wzmożonej syntezy kwasu kinureninowego oraz ksanturenowego.
Indukcja metabolizmu kinureniny/tryptofanu
Szereg mechanizmów immunologicznych i endokrynologicznych może prowadzić do oksydacyjnej indukcji metabolizmu tryptofanu (patrz ilustr. 2). Przekształcenie tryptofanu w kinureninę jest katalizowane przez dwa enzymy: przez usytuowaną w wątrobie 2,3-dioksygenazę tryptofanową (TDO) oraz przez znajdującą się przede wszystkim w makrofagach, astrocytach i w mikroglejach 2,3-dioksygenazę indolaminową (IDO). IDO jest indukowana przez prozapalne cytokiny jak interferon-γ. Mechanizmy prozapalne jak aktywowanie podstawowych czynników jądrowych (NF-KB) stres azotowy stanowią przy tym również istotne mechanizmy patologiczne (Alberti-Giani et al., 1997).
Inne cytokiny jak TNF-α i interferon-α ingerują w sposób regulujący w stopień aktywacji oraz okres trwania aktywacji ścieżki kinureninowej.
W wyniku indukcji enzymu zostaje uruchomiony wzmożony katabolizm tryptofanu ukierunkowany na metabolizm kinureniny. Wynikiem jest spadek stężenia tryptofanu w osoczu ze zredukowaną zdolnością biosyntezy serotoniny, która objawia się w obszarze przewodu pokarmowego przez zaburzenia ruchów robaczkowych oraz bolesności, zaś w obszarze centralnego układu nerwowego przez depresje i stany lękowe. Poza tym aktywacja IDO prowadzi do zwiększonej produkcji kwasu chinolinowego (QUIN) – silnego agonisty receptora NMDA (patrz ilustr. 2).
Neurotoksyczność metabolitów kinureniny
W makrofagach i mikroglejach (makrofagi centralnego układu nerwowego (ZNS)) powstaje w wyniku indukcji metabolizmu kinureniny szereg neuroaktywnych substancji, jak np. QUIN oraz hydroksykinurenina. QUIN stanowi krytyczny link między układem odpornościowym a ZNS. QUIN jest agonistą na receptorze glutaminianu typu NMDA. Aktywacja prowadzi do odczuwania bólu i stanowi klasyczny mechanizm biochemiczny typowej objawowości bólowej w infekcjach wirusowych. Jeśli receptor NMDA jest przesadnie stymulowany, prowadzi to do wewnątrzkomórkowego napływu wapnia do neuronów glutaminergicznych z degeneracją neuronów oraz znaną egzotoksycznością glutaminianową (Molz et al., 2007). Z tego patomechanizmu wywodzi się znaczenie QUIN w różnych schorzeniach neuro-zapalnych, jak np. choroba Alzheimera. Wykazano, że QUIN indukuje ekspresję IL-1 w astrozytach. IL-1 stanowi kluczowego mediatora w patogenezie choroby Alzheimera (Ting et al., 2009).
Podczas gdy QUIN stanowi agonistę na receptorze glutaminianu typu NMDA, to kinurenina działa sama jako antagonista na receptorze NMDA (patrz ilustr. 2). Oznacza to, że QUIN działa jak neurotoksyna, podczas gdy sama kinurenina wywiera skutek wspierający neurony. Trzeci metabolit – hydroksykinurenina – może już w niewielkich dawkach prowadzić do indukcji wolnych rodników a tym samym do stresu oksydacyjnego i azotowego z następstwami jakie stanowi degeneracja neuronów (Stone, 2003).
QUIN działa jak agonista NMDA czyli neurotoksyna, podczas gdy kinurenina ma działanie protekcyjne wobec neuronów jako antagonista NMDA. Hydroksykinurenina działa neurodegeneracyjnie w wyniku indukcji stresu oksydacyjnego i azotowego.
Toksyny, jak metylortęć mogą podwyższać aktywność QUIN w receptorze NMDA. W wyniku doświadczeń na zwierzętach (szczury) wykazano, że narażenie na metylortęć prowadzi do znacznej aktywacji ścieżki kinureniny i upośledza rozwój mózgu (Zanoli et al. 2001). Ftalany, stosowane na całym świecie w wyrobach plastikowych wykazują również zdolność do zwiększania wytwarzania QUIN w wyniku blokady fizjologicznego metabolizmu tryptofanu (Fukuwatari et al., 2004).
W badaniu analizowany jest poziom metabolitów tryptofanu w moczu. Tryptofan jest aminokwasem będącym substratem do produkcji serotoniniy i melatoniny, ma wpływ na funkcje wątroby, a także bierze udział w syntezie koenzymu NAD. Podwyższony poziom tryptofanu w moczu obserwujemy w zaburzeniach przemiany materii, które powodują niedobór witaminy B6 i niacyny. Zwiększone wydalanie tego związku obserwujemy także w przypadkach zwiększonej substytucji. Zaburzenia transportu aminokwasów i funkcji nerek również mogą wpłynąć na podwyższenie tego parametru w moczu.
W próbce oznaczane są;
- kwas wanilinomigdałowy,
- kwas homowanilinowy,
- kwas 5-hydroksyindolooctowy,
- tryptofan,
- kwas ksanturenowy,
- L-kinurenina,
- kwas kinureninowy,
- kwas chinolinowy.
Obliczany jest także stosunek L-kinurenina/tryptofan, a także stosunek kwasu kinureninowego/L-kinureniny.
Acetylokarnityna
System karnitynowy umożliwia transport aktywnych metabolicznie kwasów tłuszczowych poprzez błonę mitochondrialną do miejsc beta-oksydacji w formie karnityzowanych kwasów tłuszczowych i tym samym stanowi ważny element systemu energetycznego komórki.
W procesie tym ważne są 4 enzymy:
Acetylotransferaza karnitynowa
– enzym znajdujący się w mitochondriach i peroksysomach, który przenosi krótkołańcuchową grupę acylową z koenzymu A na karnitynę usuwając toksyczne grupy acylowe z mitochondriów
Oktanylotransferaza karnitynowa
transport średnio łańcuchowych reszt acylowych z peroksysomów do mitochondriów
Palmitoilotransferaza karnitynowa
transportująca długołańcuchowe kwasy tłuszczowe do mitochondriów
Mitochondrialna acetylotransferaza
karnitynowa zlokalizowana w wewnętrznych błonach mitochondrialnych umożliwiająca wzajemna wymianę acylokarnityny i karnityny poprzez błony mitochondrium w obydwu kierunkach.
Niskie wartości acetylo-L-karnityny wskazują pośrednio na zmniejszona aktywność acetylotransferazy karnitynowej, jak i zmniejszony poziom L-karnityny zgodnie z wzorem:
L-karnityna + AcCoA -> Acetyl-L-karnityna + CoA
Niedobory L-Karnityny i Acetyl-L-Karnityny prowadzą do zmniejszonej zdolności komórek do generowania energii pochodzącej z utleniania kwasów tłuszczowych i tym samym do zmniejszenia tworzenia Acetylokoenzymu A i zależnych od niego procesów metabolicznych. Kwasy tłuszczowe mogą przy tym ulegać przemianom w cholesterol lub inne glicerydy co prowadzić może np. do tworzenia blaszek miażdżycowych.
Glutation (zredukowany/utleniony)
Glutation (GSH) jest peptydem mającym silne zdolności antyoksydacyjne. Uczestniczy on w wielu procesach ochronnych. Oznaczenie całkowitego glutationu w odniesieniu do hemoglobiny daje pogląd na zdolności samoobronne organizmu. Przy wielu chorobach przebiegających ze stresem azotowym i tlenowych pojawia się on w formie oksydowanej (GSSG). Określenie stosunku GSH/GSSG daje pogląd na przebieg procesów antyoksydacyjnych.