Artykuł został zaktualizowany 25.02.2025 r.
- Jak dochodzi do zakażenia wirusem brodawczaka ludzkiego (HPV) u mężczyzn?
- HPV – objawy u mężczyzny
- Kłykciny kończyste – przyczyny, objawy, diagnostyka, leczenie
- Rak prącia – przyczyny, objawy, diagnostyka, leczenie
- Testy HPV dla mężczyzn. Jak wygląda badanie HPV u mężczyzn?
- Profilaktyka zakażeń wirusem brodawczaka ludzkiego (HPV) – szczepienia przeciwko HPV u chłopców
Wirus brodawczaka ludzkiego (HPV, Human Papilloma Virus) to główna przyczyna powstawania nowotworu szyjki macicy u kobiet. Warto jednak mieć świadomość, iż infekcje HPV występują również u mężczyzn i mogą mieć równie poważne skutki, doprowadzając do zachorowania na raka jamy ustnej lub prącia.
Wirus brodawczaka ludzkiego (HPV) to rodzina ok. 200 typów wirusów, wśród których znajdują się rodzaje wysokoonkogenne, zwiększające ryzyko zachorowania na nowotwór (szyjki macicy, prącia, sromu, odbytu, jamy ustnej) oraz nieonkogenne, powodujące powstawanie tzw. kłykcin kończystych. Te ostatnie nie są groźne w skutkach, jednak świadczą o występowaniu w organizmie infekcji wirusem brodawczaka ludzkiego (HPV).
Jak dochodzi do zakażenia wirusem brodawczaka ludzkiego (HPV) u mężczyzn?
Zakażenie wirusem brodawczaka ludzkiego (HPV) jest najczęściej infekcją przenoszoną drogą płciową, można się zarazić podczas stosunku seksualnego (zarówno pochwowego, jak i analnego lub oralnego). Niestety równie groźny w skutkach może być kontakt bezpośredni z wydzielinami nosiciela (ślina) lub skórą (krocze, pachwiny, odbyt). Z tego powodu prezerwatywy poważnie ograniczają ryzyko zachorowania, ale nie chronią przed nim w 100%, bowiem wirus może być obecny poza obszarem ochrony.
Wirus HPV zakaża komórki naskórka – wielowarstwowego nabłonka, stanowiącego najbardziej powierzchowną, zewnętrzną warstwę skóry. Atakuje komórki podstawne (ułożone w jednym rzędzie komórki o kształcie walca), w których ciągle zachodzą podziały komórkowe, dzięki czemu naskórek może się odbudowywać. Dlatego, jeśli namnażanie (replikacja) wirusa ma niewielkie nasilenie, infekcja sama się wygasza. Jednak w miarę dojrzewania i różnicowania się zakażonych komórek replikacja nasila się, a zainfekowane komórki ulegają transformacji i powstają charakterystyczne dla wirusa zmiany. Namnażaniu się wirusa brodawczaka ludzkiego (HPV) sprzyjają zaburzenia odporności.
Więcej o wirusie HPV przeczytasz TUTAJ.
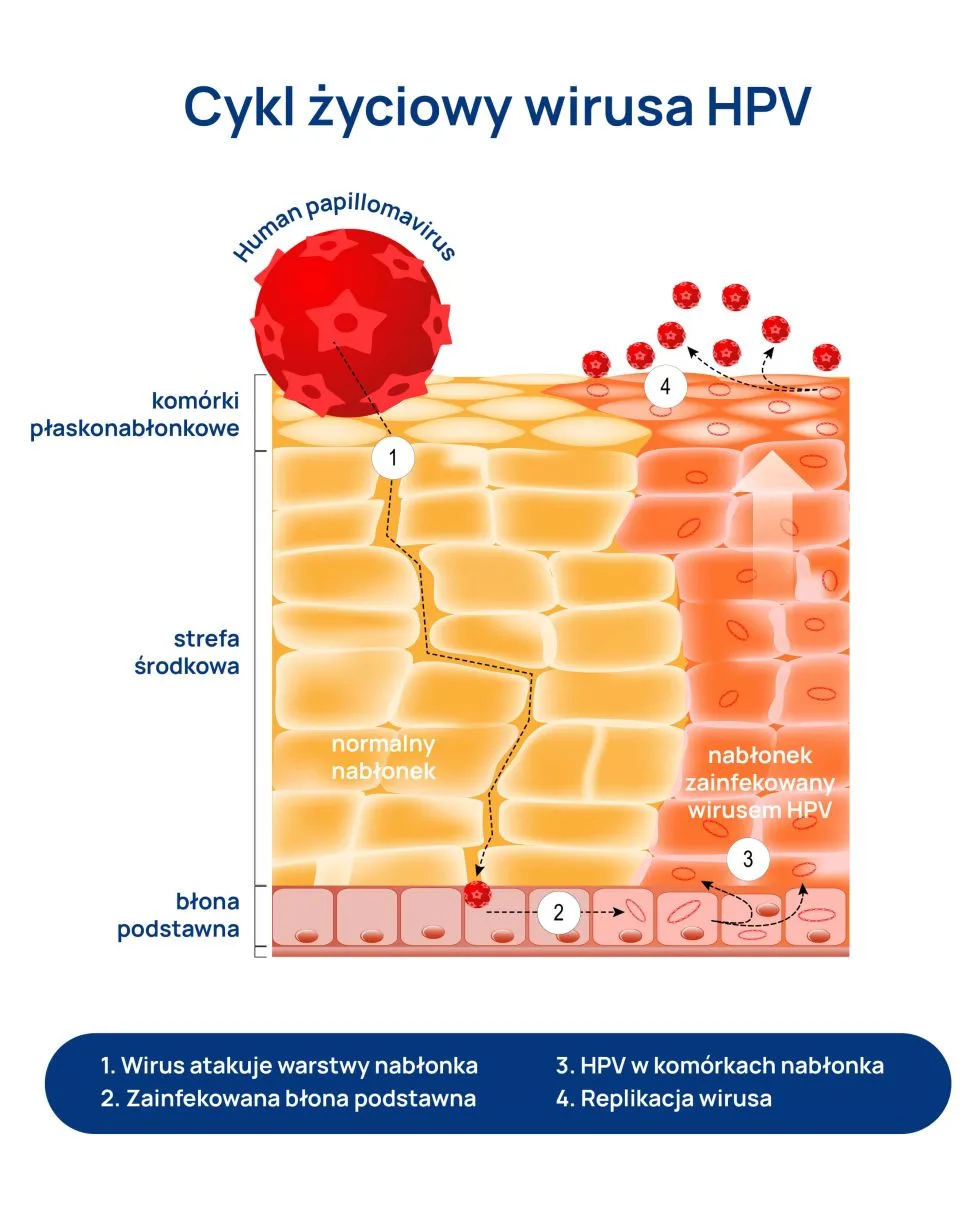
Infekcja wirusem brodawczaka ludzkiego (HPV) dotyka najczęściej mężczyzn pomiędzy 18. a 39. rokiem życia, chociaż możliwa jest u każdego aktywnego seksualnie mężczyzny. Konsekwencje zakażenia mogą się również pojawiać w ciągu całego życia, np. rak prącia spotykany jest zazwyczaj u mężczyzn 60+.
Statystyki mówią, iż ok. 80% populacji (zarówno mężczyzn, jak i kobiet) było zakażone wirusem brodawczaka ludzkiego (HPV) przynajmniej raz w życiu.
Możliwe jest także zakażenie wskutek zaniedbań higienicznych, np. poprzez używanie wspólnego ręcznika.
HPV – objawy u mężczyzny
Infekcja wirusem HPV u mężczyzny może przebiegać bezobjawowo i – jak napisano wyżej – jeśli jest niewielka sama się wygasza.
Infekcja HPV u mężczyzny może przebiegać pod postacią:
- kłykcin kończystych,
- raka odbytu,
- raka gardła,
- raka prącia.
Objawy HPV u mężczyzny mogą występować jako:
- grudkowane wykwity na prąciu, w okolicach cewki moczowej lub odbytu – kłykciny kończyste
- pieczenie, świąd w okolicy odbytu, guzek, śluz/świeża krew w kale – rak odbytu
- uczucie przeszkody w gardle, problemy z połykaniem, krztuszenie się, uporczywy kaszel – możliwe symptomy raka gardła
- płaski wykwit na napletku lub żołędzi, powiększający się w czasie, czasem uwypukla się lub wrzodzieje – możliwe symptomy raka prącia.
Więcej o raku odbytu przeczytasz TUTAJ. Kłykciny kończyste i rak prącia opisane są poniżej.
Kłykciny kończyste – przyczyny, objawy, diagnostyka, leczenie
Kłykciny kończyste to jedna z najczęstszych chorób przenoszonych drogą płciową. Przyczyną ich powstania jest infekcja nieonkogennymi typami wirusa brodawczaka ludzkiego (HPV) – 6 i 11. Oznacza to, iż kłykciny są zmianami, które nie przekształcą się w nowotwór, chociaż – jeśli występują w dużym nasileniu – mogą utrudniać funkcjonowanie i być przyczyną problemów związanych ze współżyciem (pacjenci wycofują się z aktywności seksualnej) oraz zdrowiem psychicznym (obniżenie poczucia własnej wartości, obawy związane z płodnością oraz zachorowaniem na nowotwór).
Diagnostyka kłykcin kończystych nie wymaga badań laboratoryjnych, chociaż można pobrać wymaz z żołędzi penisa, aby potwierdzić, że zakażenie wywołane jest tylko wirusami nieonkogennymi. Może zdarzyć się sytuacja, gdy źródłem infekcji jest zakażenie mieszane.
Okres inkubacji choroby – czyli czas od momentu zakażenia do pojawienia się charakterystycznych objawów klinicznych – jest zmienny, trwa od kilku do kilkunastu miesięcy. W przypadku mężczyzn kłykciny kończyste zlokalizowane są najczęściej na prąciu (na wewnętrznej blaszce napletka, na brzegu żołędzi lub w okolicach wędzidełka), rzadziej w okolicach cewki moczowej lub odbytu. Są to charakterystyczne grudkowate wykwity, często liczne. Mogą samoistnie zanikać, ale mają skłonność do nawracania. Na ogół nie powodują żadnych dolegliwości, czasem pacjent odczuwa świąd.
Rozpoznanie kłykcin opiera się na charakterystycznym obrazie klinicznym. Pacjenci z kłykcinami powinni wykonać badania przesiewowe w kierunku innych chorób przenoszonych drogą płciową (HIV, kiła, chlamydioza, rzeżączka, WZW typu B i C).
Leczenie kłykcin nie jest przyczynowe, nie opracowano leku zwalczającego zakażenie wirusem brodawczaka ludzkiego (HPV). U osób, które mają niewielkie zmiany, można zastosować leczenie farmakologiczne, większe i rozległe zmiany usuwa się w sposób inwazyjny (łyżeczkowanie, wycięcie, kriochirurgia, laseroterapia).
Rak prącia
Rak prącia jest rzadkim nowotworem, w Polsce występuje u ok. 200 mężczyzn rocznie. Typowy pacjent ze zmianą nowotworową tego organu ma ponad 60 lat, jednak zawsze należy pamiętać, że możliwe jest jego wystąpienie również u młodszych osób.
Jednym z podstawowych czynników ryzyka przyczyniających się do powstawania raka prącia jest brak higieny osobistej. W kulturach krajów, gdzie praktykowane jest obrzezanie chłopców, nowotwór prącia diagnozowany jest rzadziej. Tłumaczy się to faktem, iż usunięcie napletka ułatwia zachowanie higieny.
Do powstawania tego schorzenia przyczynia się również wirus brodawczaka ludzkiego (HPV) – wysokoonkogenny typ 16. Poza tym czynnikami sprzyjającymi są współistnienie zakażenia HIV, duża aktywność seksualna, stulejka i przewlekłe stany zapalne.
Pierwszym objawem nowotworu prącia, który najczęściej zauważa pacjent, jest płaska, czasem wrzodziejąca zmiana umiejscowiona na napletku lub na żołędzi. Zmiana może na początku przypominać plamkę, jedna rośnie i uwypukla się wraz z upływem czasu. Wszystkie objawy, które niepokoją, powinny być skonsultowane z lekarzem. Najczęściej nie są to zmiany w typie nowotworu, jednak warto zachować ostrożność.
W przypadku raka prącia atakowane są również węzły chłonne w pachwinach – obecność powiększonych guzków jest dla niektórych pacjentów powodem do zgłoszenia się do lekarza.
Diagnostyka nowotworu prącia zaczyna się od wywiadu oraz badania lekarskiego. O rozpoznaniu rozstrzyga biopsja. Dodatkowo lekarz zleci USG jamy brzusznej czy RTG klatki piersiowej, w celu oceny występowania przerzutów, może to być również tomografia komputerowa czy rezonans magnetyczny.
Wcześnie wykryty nowotwór prącia, w którym nie ma przerzutów, rokuje dobrze, a metody leczenia oszczędzają narząd (mogą to być krioterapia, laseroterapia, brachyterapia czy usunięcie samej żołędzi). W stanach bardziej zaawansowanych leczenie jest często radykalne i polega na częściowej lub całkowitej amputacji prącia, czyli tzw. penektomii.
Testy HPV dla mężczyzn. Jak wygląda badanie HPV u mężczyzn?
Wykrywanie obecności wirusa brodawczaka ludzkiego (HPV) u mężczyzn możliwe jest po pobraniu wymazu z miejsc chorobowo zmienionych na penisie lub z cewki moczowej.
Jak wygląda badanie HPV u mężczyzn? Materiał pobiera się z żołędzi penisa, najlepiej z miejsc chorobowo zmienionych – jeśli jest to możliwe, ponieważ zakażenie u mężczyzn często przebiega bezobjawowo. Próbkę pobiera się specjalną wymazówką, poprzez kilkukrotne pocieranie zmiany chorobowej ruchem spiralnym. Jeśli badanie wykonywane jest w celach profilaktycznych i nie ma widocznej zmiany chorobowej, materiał należy pobrać z rowka zażołędnego wokół penisa, na całej jej długości.
Wymaz można również pobrać z cewki moczowej, w tym celu używa się tzw. mini wymazówki, którą wprowadza się na ok. 1 cm do cewki moczowej, przytrzymuje 5-10 sekund, a następnie kilkakrotnie ją obraca.
W przypadku badań w kierunku wirusa brodawczaka ludzkiego (HPV) ważne jest przygotowanie pacjenta do badania.
- 48 godzin przed pobraniem wymazu nie należy stosować żadnych leków, zwłaszcza w postaci maści,
- 24 godziny przed pobraniem należy wstrzymać się od współżycia płciowego oraz ograniczyć zabiegi higieniczno-pielęgnacyjne okolic narządów płciowych.
Istnieje kilka typów testów, wykrywających różne typy wirusów brodawczaka ludzkiego (HPV) w różnych konfiguracjach.
- HPV-GEN – Test różnicujący 32 genotypy wirusa HPV, w tym wysokoonkogenne oraz nieonkogenne (6, 11, 16, 18, 26, 31, 33, 35, 39, 40, 42, 43, 44, 45, 51, 52, 53, 54, 56, 58, 59, 61, 62, 66, 67, 68, 70, 73, 81, 82, 83, 89)
- HPV-G41 – Test wykrywający i różnicujący 41 typów wirusa HPV (6, 11, 16, 18, 26, 31, 33, 35, 39, 40, 42, 43, 44, 45, 51, 52, 53, 54, 55, 56, 58, 59, 61, 62, 64, 66, 67, 68a, 68b, 69, 70, 71, 72, 73, 81, 82 (IS39), 83, 84, 87, 89 (CP6108), 90)
- HPV-HR – Test wykrywający DNA 14 wysokoonkogennych (mogących być przyczyną raka) typów wirusa (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59)

Profilaktyka zakażeń wirusem brodawczaka ludzkiego (HPV) – szczepienia przeciwko HPV u chłopców
Wirus brodawczaka ludzkiego (HPV) to czynnik sprawczy groźnych nowotworów, zarówno u kobiet, jak i u mężczyzn. Rak szyjki macicy – dzięki programowi badań przesiewowych – może być wykryty wcześniej, natomiast pozostałe nowotwory HPV-zależne (gardło, prącie, odbyt) nie mają takich programów wczesnego wykrywania. Dlatego ważnym elementem profilaktyki na dziś jest szczepionka przeciwko HPV.
W Polsce dostępne są trzy typy szczepionek, a od 1 czerwca 2023 bezpłatnymi szczepieniami objęte są dziewczynki i chłopcy w wieku 12 i 13 lat. Poza refundacją zaszczepić się może również młodzież po 14 r.ż. i dorośli. Więcej o szczepionce przeczytasz TUTAJ.
Piśmiennictwo
- Kongres Interdyscyplinarny „Najnowsze trendy w profilaktyce, diagnostyce i leczeniu raka szyjki macicy”, wydarzenie hybrydowe, 3.03.2023.
- Konferencja online „Profilaktyka, diagnostyka i leczenie chorób związanych z HPV w ginekologii i ginekologii onkologicznej”, 10-11 maja 2024.