W wątrobie zachodzi wiele złożonych procesów, powiązanych z funkcjonowaniem innych narządów i układów:
odgrywa kluczową rolę w przemianach białek – w wątrobie powstają białka osocza, wytwarzane są aminokwasy i inne związki,
jest miejscem metabolizmu węglowodanów – w wątrobie zachodzi magazynowanie, wytwarzanie i uwalnianie zasobów glukozy w zależności od potrzeb danego organizmu,
pełni kluczową rolę w gospodarce tłuszczowej organizmu – odbywa się w niej przekształcanie węglowodanów i białek w tłuszcze, synteza lipoprotein, fosfolipidów i cholesterolu,
w wątrobie zachodzi detoksykacja związków endogennych np. hormonów steroidowych oraz egzogennych np. leków, toksyn, które docierają do tego narządu z jelit drogą krążenia wrotnego,
jest miejscem syntezy 25-hydroksycholekalcyferolu,
w wątrobie gromadzone są zapasy witamin A, D, B12 i żelaza w postaci ferrytyny,
pełni bardzo ważne funkcje immunologiczne.
Co ciekawe, wątroba jest największym gruczołem ustrojowym w ciele człowieka, u dorosłego człowieka jej waga osiąga około 1,5 kg.
Przewlekłe choroby wątroby mogą prowadzić do stopniowego i przeważnie nieodwracalnego uszkodzenia tego narządu. Następstwem jest zaburzenie funkcjonowania wątroby zależnie od stopnia zniszczenia jej komórek. Do głównych chorób wątroby należą ostre i przewlekłe zapalenie wątroby, marskość wątroby, choroba stłuszczeniowa wątroby i toksyczne uszkodzenie wątroby.
Żywienie osób z przewlekłymi chorobami wątroby zawsze indywidualnie dostosowujemy do stanu zdrowia pacjenta. Powinno uwzględniać:
stan odżywienia chorego,
choroby współistniejące,
wydolność wątroby,
występujące zaburzenia metaboliczne.
U pacjentów z przewlekłymi chorobami wątroby, u których nie rozpoznajemy cech niewydolności narządu, którzy dobrze tolerują normalną dietę, nie są konieczne dodatkowe restrykcje dietetyczne. Ważne, aby dieta składała się z urozmaiconych posiłków, produktów dozwolonych.
Posiłki powinny być jedzone regularnie w odstępach 3 h. Śniadanie zjadamy do 1 h po przebudzaniu a kolację na 2-3 h przed pójściem spać. Dziennie pacjent powinien zjadać 5-6 odpowiednio zbilansowanych posiłków, dostosowanych do indywidualnych potrzeb danej osoby, odpowiedniej wartości energetycznej diety i podaży białka, aby zapobiec niedożywieniu. U pacjentów, którzy dobrze tolerują tłuszcze, nie ogranicza się znacznie ich ilości, natomiast u osób źle tolerujących, redukuje się poziom tłuszczy do ilości dobrze przyswajalnych. Jeżeli mamy do czynienia z brakiem apetytu u pacjenta, nudnościami, biegunkami czy innymi objawami powodującymi niechęć do spożywania posiłków, pokarmy powinny być wprowadzane częściej, w ograniczonych objętościowo porcjach. Zbyt obfite posiłki mogą powodować dolegliwości dyspeptyczne.
Wszystkie posiłki powinny być spożywane w spokojnej atmosferze. Należy długo przeżuwać każdy kęs, nie rozmawiamy zbyt ekspresywnie przy jedzeniu, aby nie połykać powietrza. Dania nie mogą być bardzo gorące ani zbyt zimne. Posiłki powinny być przyrządzane w dniu spożycia, powinny być świeże i zachęcać sposobem podania do zjedzenia. Nie powinno się ich odgrzewać lub długo przechowywać w lodówce. Sposób przyrządzania to gotowanie, na parze, pieczenie, grillowanie na patelni grillowej. Niedozwolone jest przesmażanie, smażenie w tłuszczach czy przyrządzanie pokarmów w panierkach, duszenie.
Do picia zalecana jest woda mineralna niegazowana – 1,5 l na dobę, można też przyrządzać słabe napary herbaty czarnej bez cukru lub herbatki owocowe, kompoty ze świeżych, dozwolonych owoców bez cukru. Napoje należy przyjmować między posiłkami. Nie powinno się popijać podczas jedzenia.
Przykładowy jadłospis w chorobach wątroby
I śniadania:
bułka kukurydziana posmarowana cienko masłem z chudą wędliną drobiową, sałatą dowolną i pomidorem sparzonym bez skórki,
płatki owsiane (zalane wrzątkiem, aby wypłukać z płatków szczawiany, po kilku minutach odlewamy wrzątek) dodajemy owoce maliny/borówkę amerykańska/jabłko bez skórki (owoce mogą być lekko podgotowane w malutkiej ilości wody) plus jogurt naturalny z niską zawartością tłuszczu,
omlet z samych białek jaj i mąki pszennej oraz mleka 2%, pieczemy bez dodatków przygotowany bez tłuszczu na patelni teflonowej, możemy użyć papieru do pieczenia. Podajemy z dżemem malinowym/truskawkowym/morelowym – najlepiej własnej roboty.
II śniadania:
pieczywo pszenne posmarowane cienko masłem z twarożkiem z sera chudego, z dodatkiem jogurtu naturalnego o niskiej zawartości tłuszczu z odrobina soli, do tego ogórek zielony,
ryż jaśminowy ugotowany z łososiem upieczonym, pokrojonym w plasterki do tego sałata, pomidor bez skórki i ogórek zielony, kropimy oliwą z oliwek i kilkoma kroplami cytryny, przyprawiamy do smaku dozwolonymi ziołami.
Obiady:
udka z kurczaka doprawione ziołami i przyrządzone na parze, podane z ziemniakami purée, do tego buraczki na ciepło,
zupa krupnik, na piersi kurczaka bez skórki, do tego warzywa starte marchew/pietruszka/seler, ziemniaki i kasza jęczmienna drobna, można na talerzu doprawić naturalnym jogurtem z niską zawartością tłuszczu,
dorsz/miruna/mintaj pieczony w folii z odrobiną masła i z koperkiem, do tego ryż jaśminowy lub biały, gotowany kalafior pokropiony oliwą z oliwek.
Podwieczorki:
pieczone warzywa – marchew/pietruszka obrane, pokrojone w słupki i cukinia obrana, pokrojona w grubsze plastry, doprawione dozwolonymi ziołami, pieczemy w piekarniku 180C ok. 20 minut,
serek wiejski z pomidorem bez skórki i pieczywo pszenne/kukurydziane/ryżowe,
zupa krem z brokuła ugotowana na wywarze warzywnym podana z grzankami z chleba pszennego.
Kolacje:
placuszki ziemniaczane z purée ziemniaczanego, upieczone w piekarniku i podane z jogurtem naturalnym o niskiej zawartości tłuszczu, do tego ogórek zielony bez skóry,
pieczywo pszenne z wędliną własnej roboty z udźca indyka, do tego pomidor bez skóry i dowolna sałata,
sałatka z makaronu ryżowego/pszennego do tego upieczona w ziołach pierś z kurczaka pokrojona w paseczki, dowolna sałata, pomidorki koktajlowe i ugrillowana lub upieczona w piekarniku cukinia bez skórki, wszystko kropimy sosem z oliwy z oliwek, kilkoma kroplami cytryny i ulubionymi, dozwolonymi ziołami.
Piśmiennictwo
Włodarek D., Lange E., Kozłowska L., Głąbska D., Dietoterapia. Wydawnictwo Lekarskie PZWL, Warszawa 2014
Ciborowska H., Rudnicka A., Dietetyka. Żywienie zdrowego i chorego człowieka. Wydawnictwo Lekarskie PZWL, Warszawa 2015.
Gertig H., Przysławski J., Bromatologia. Zarys nauki o żywności i żywieniu. Wydawnictwo Lekarskie PZWL,
Dr n.med. D. Waśko – Czopnik, Dieta wątrobowa, http://waskoczopnik.pl/pdf/Dieta_watrobowa.pdf
Witamina B1 (tiamina) to jedna z rozpuszczalnych w wodzie, witamin grupy B. Ciekawostką jest fakt, że została zidentyfikowana jako pierwszy związek w tej grupie, dlatego została oznaczona cyfrą jeden, a odkrycia dokonał polski biochemik Kazimierz Funk, prowadząc badania nad przyczynami choroby beri-beri. Jemu również zawdzięczamy wprowadzenie powszechnie dziś używanego terminu „witamina”, czyli substancja niezbędna do życia (z łacińskiego „vita”), zawierająca w swojej strukturze azot (należąca do grupy amin).
Witamina B1 (tiamina) – rola w organizmie
Witamina B1 (tiamina) jest składnikiem wielu enzymów, odgrywając istotną rolę w przemianach prowadzących do uzyskania energii, zwłaszcza w przemianach węglowodanów.
Uczestniczy również w przewodnictwie nerwowym, syntezie nukleotydów, oraz procesach oddychania komórkowego. Dzięki temu obserwuje się jej korzystny wpływ na układ nerwowy, mięśniowy i sercowo-naczyniowy. U osób nieprzystosowanych do wysiłku fizycznego uczestniczy w usuwaniu nadmiaru kwasu pirogronowego. Tiamina wywiera hamujący wpływ na esterazę choliny, co skutkuje zwiększoną aktywnością acetylocholiny, dlatego niedobór witaminy B1 przekłada się na zwolnienie perystaltyki jelit i zmniejszenia napięcia mięśni gładkich.
Witamina B1 (tiamina) w postaci difosforanu tiaminy jest koenzymem trzech kompleksów enzymatycznych:
dehydrogenazy α-ketoglutaranowej – cykl kwasu cytrynowego (cykl Krebsa),
dehydrogenazy ketokwasów uczestniczących w metabolizmie aminokwasów leucyny, izoleucyny i waliny.
Tiamina w postaci trifosforanu tiaminy uczestniczy w przewodnictwie nerwowym.
Biodostępność witaminy B1 (tiaminy) – czynniki wpływające na wchłanianie
Witamina B1 (tiamina) wchłania się w jelicie cienkim, w wyniku transportu aktywnego. Wolna postać tiaminy występująca w błonie śluzowej jelita cienkiego ulega przekształceniu w związek aktywny – pirofosforan.
Duże dawki witaminy B1 (tiaminy) dostarczane z pokarmem lub suplementacją, powodują:
szybkie wysycenie mechanizmów transportowych i ograniczenie wchłaniania,
wysycenie tkanek docelowych,
nasilenie wydalanie witaminy B1 z moczem.
Dlatego suplementacja tiaminy w przypadku, gdy w organizmie nie występują niedobory, jest bezzasadna.
Witamina B1 (tiamina) jest stabilna w środowisku kwaśnym i obojętnym, w którym nie jest wrażliwa na ogrzewanie nawet do 100 stopni Celsjusza. Natomiast takie same warunki w środowisku zasadowym niszczą witaminę B1. Tiamina jest również wrażliwa na działanie kawy i herbaty, które ograniczają jej wchłanianie.
Zapotrzebowanie i źródła witaminy B1 (tiaminy) w diecie
Witamina B1 (tiamina) zawarta jest zarówno w produktach roślinnych, jak i pochodzenia zwierzęcego. Biorąc pod uwagę udział poszczególnych grup produktów, w całodziennej diecie najwięcej witaminy B1 dostarczają:
Zapasy witaminy B1 (tiaminy) w organizmie człowieka nie są duże. Jej brak objawia się już po ok. trzech tygodniach niedoborów. Początkowe objawy braku witaminy B1 to zmęczenie, drażliwość, zaburzenia nastroju, zaburzenia koncentracji. Dłużej trwające niedobory mogą wywoływać objawy podobne do tych, które obserwuje się w takich chorobach układu nerwowego jak choroba Alzheimera, Parkinsona, Huntingtona lub Wernicke-Korsakoffa. Niedobór tiaminy powoduje zaburzenia gospodarki węglowodanowej, a mózg czerpie energię prawie wyłącznie z tej grupy produktów, dlatego zmiany czynnościowe w ośrodkowym układzie nerwowym pojawiają się stosunkowo szybko, jeśli niedobór trwa dłużej, może dochodzić do rozpadu i zaniku osłonki mielinowej nerwów.
Niedobór witaminy B1 (tiaminy) to również objawy ze strony przewodu pokarmowego – zaburzenia perystaltyki jelit, zaburzenia wydzielania soku żołądkowego, a co za tym idzie, zaburzenia trawienia i przyswajania składników pokarmowych.
Najpoważniejszym skutkiem niedoboru witaminy B1 (tiaminy) jest choroba beri-beri charakteryzująca się zmianami degeneracyjnymi w układzie nerwowym i zanikiem mięśni szkieletowych. Inne objawy to zaburzenia sercowo-naczyniowe i obrzęki.
Nadmierna suplementacja witaminy B1, gdy nie stwierdza się jej niedoboru, również jest niekorzystna.
Piśmiennictwo
Ciborowska H., Rudnicka A., Dietetyka. Żywienie zdrowego i chorego człowieka. Wydawnictwo Lekarskie PZWL, Warszawa 2015.
Gertig H., Przysławski J., Bromatologia. Zarys nauki o żywności i żywieniu. Wydawnictwo Lekarskie PZWL, Warszawa 2015.
Triglicerydy (trójglicerydy), potocznie nazywane tłuszczami, są to lipidy, których cząsteczka jest zbudowana z połączenia z glicerolu i trzech łańcuchów kwasów tłuszczowych. Długość tych łańcuchów wynosi od 14 do 24 atomów węgla i może zawierać różną liczbę podwójnych wiązań. Biologiczne właściwości triglicerydów będą zależały od tego, jakie kwasy tłuszczowe będą budowały cząsteczkę. Rolą triglicerydów w organizmie jest dostarczanie, jak i przechowywanie energii (w tkance tłuszczowej), jak również transport kwasów tłuszczowych (do celów innych niż wykorzystanie energetyczne).
Metabolizm triglicerydów
Gdy człowiek zjada tłuszcze, te trafiają do jelit, gdzie po trawieniu są wchłanianie przez komórki wyściełające jelita, zwane enterocytami. W enterocytach produkty trawienia tłuszczu są przekształcane ponownie w triglicerydy, a następnie umieszczane w chylomikronach – dużych gęstych lipoproteinach będących przenośnikiem triglicerydów. Chylomikrony są następnie wydobywane z enterocytów i trafiają do układu limfatycznego, który przenosi je do wątroby i układu krwionośnego. Dalej mogą one albo zostać wchłonięte przez wątrobę, albo ulegać rozkładowi tłuszczu na powierzchni naczyń krwionośnych przez enzym zwany lipazą lipoproteinową (LPL). W wątrobie triglicerydy są następnie „przepakowane” w inną formę lipoprotein, znane jako VLDL (z ang. Very-Low-Density Lipoproteins – VLDL). VLDL są potem transportowane do innych części ciała przez układ krążenia. W tkankach obwodowych VLDL ulega przemianom – triglicerydy są z niego usuwane przez LPL, a pozostała część tworzy IDL (z ang. Intermediate-Density Lipoproteins – IDL). IDL mogą zostać dalej wychwycone przez wątrobę lub przekształcone w LDL (z ang. Low-Density Lipoproteins – LDL).
Ponieważ 80% wszystkich triglicerydów znajduje się w tzw. lipoproteinach bogatych w triglicerydy (VLDL i IDL), stężenie triglicerydów będzie odpowiadało liczbie tych lipoprotein, a w efekcie ryzyku miażdżycy związanemu z ich występowaniem.
Należy pamiętać, że ilość triglicerydów przenoszona przez VLDL i IDL jest zmienna i nie zawsze odpowiada dokładnie liczbie tych lipoprotein. Stąd też pomocny okazuje się pomiar stężenia cholesterolu nie-HDL, który łączy w sobie informację na temat zarówno VLDL/IDL, jak również LDL będącego główną lipoproteiną odpowiedzialną za rozwój miażdżycy.
Jak przygotować się do badania triglicerydów?
Pomiar stężenia triglicerydów wykonuje się najczęściej w ramach profilu lipidowego, więc badanie nie jest związane ze specjalnymi przygotowaniami. Tradycyjnie przyjmowano, że oceny profilu lipidowego należy dokonać po 10-12 godzinnym okresie niespożywania posiłków (na czczo). Jednak z racji, iż organizm przez większość doby nie jest na czczo, a profil lipidowy ma odzwierciedlać rzeczywisty stan gospodarki lipidowej, obecnie nie wymaga się pobierania krwi na lipidogram na czczo. Poposiłkowe stężenie triglicerydów jest średnio ~27 mg/dL (0,3 mmol/L) wyższe od stężenia na czczo. Stąd też pomiar pobrany po spożyciu posiłku jest ciągle wiarygodnym parametrem, a taki sposób badania jest wygodniejszy dla pacjenta. Należy jednak pamiętać o zmienności stężenia triglcerydów względem liczby lipoprotein – ta zmienność jest większa po posiłku. Stąd też w wypadku uzyskania stężenia triglicerydów nie na czczo >400 mg/dL, warto powtórzyć badanie na czczo.
Hipertriglicerydemia
Hipertriglicerydemią nazywamy stan, w którym stężenie triglicerydów:
Na czczo wynosi >150 mg/dL,
Nie na czczo wynosi > 175 mg/dl.
Dodatkowo hipertriglicerydemię można podzielić na:
Hipertriglicerydemię umiarkowaną – jeśli stężenie nie przekracza 500 mg/dL.
Hipertriglicerydemię ciężką – jeśli stężenie przekracza 500 mg/dL, przy czym alarmujące jest stężenie >880 mg/dL, mogące sugerować genetyczne podłoże hipertriglicerydemii.
Przyczyny hipertriglicerydemii mogą być zarówno pierwotne, jak i wtórne.
Do pierwotnych przyczyn należy wieloczynnikowa hipertriglicerydemia (wielogenowo dziedziczne podwyższone stężenie triglicerydów), złożona hiperlipoproteinemia (wielogenowo dziedziczne podwyższone stężenie triglicerydów i cholesterolu LDL) oraz bardzo rzadkie przypadki rodzinnej chylomikronemii oraz dysbetalipoproteinemii.
Do wtórnych przyczyn należą: nadmierna masa ciała oraz zespół metaboliczny, nadmierne spożycie alkoholu, dieta bogata w proste węglowodany, choroby nerek, ciąża (zwłaszcza III trymestr), niedoczynność tarczycy, a także leki (glikokortykosteroidy, doustne estrogeny, β-blokery, diuretyki tiazydowe, retinoidy). Ponieważ większość przypadków hipertriglicerydemii jest umiarkowana, prawdopodobnie będzie ona związana z nałożeniem podłoża wielogenowego z dodatkowymi czynnikami wtórnymi.
W umiarkowanej hipertriglicerydemii głównym skutkiem zdrowotnym będzie efekt obecności VLDL i LDL na tworzenie się blaszki miażdżycowej, zwiększający ryzyko sercowo-naczyniowe. Ponieważ LDL jest główną lipoproteiną odpowiedzialną za rozwój miażdżycy, wkład ze strony VLDL i LDL będzie mniejszy. Dlatego też w umiarkowanej hipertriglicerydemii większy nacisk będzie kładziony na stężenie cholesterolu LDL. Z tego względu, w zależności od wyjściowego ryzyka sercowo-naczyniowego, lekarz może zdecydować o włączeniu leczenia statynami, które poza obniżeniem cholesterolu LDL obniżają również stężenie triglicerydów. W przypadku, gdy osiąga się docelowe stężenie cholesterolu LDL, a stężenie triglicerydów są ciągle podwyższone, można zastosować leki z grupy fibratów lub kwasy tłuszczowe omega-3.
Ciężka hipertriglicerydemia jest związana nie tylko ze zwiększoną liczbą VLDL i IDL we krwi, ale również chylomikronów, które znacznie zwiększają ryzyko ostrego zapalenia trzustki – to właśnie szybkie działanie terapeutyczne ma ograniczyć ryzyko tego zdarzenia. Dlatego celem jest ograniczenie liczby chylomikronów, VLDL i LDL, stąd w pierwszej kolejności zależy nam na obniżeniu stężenia triglicerydów. Głównymi lekami w tym przypadku są fibraty, kwas tłuszczowe omega-3 oraz niacyna.
Niezależnie od postaci hipertriglicerydemii zmiana stylu życia jest kluczowa dla powodzenia leczenia. Zredukowanie nadmiernej masy ciała jest jednym z najbardziej efektywnych środków w kontrolowaniu hipertriglicerydemii. Ograniczenie spożycia alkoholu jest równie ważne – nawet umiarkowane ilości mogą poważnie wpłynąć na poziomy triglicerydów. Regularna aktywność fizyczna jest także niezwykle istotna, ponieważ sprzyja utracie masy ciała, ale ma również bezpośredni wpływ na obniżenie triglicerydów. Korzystny wpływ ma również podaż kwasów tłuszczowych omega-3 w diecie, np. ryby, orzechy. Ograniczenie spożycia mono- i disacharydów (cukrów prostych) oraz zastąpienie tłuszczów nasyconych (np. masło) tłuszczami mono- lub wielonienasyconymi (np. oliwa z oliwek) są mniej efektywne, ale wciąż wartościowe w kontekście ogólnego zarządzania poziomem triglicerydów.
Hipotriglcerydemia
Hipotryglicerydemia, czyli stan bardzo niskiego poziomu triglcerydów we krwi, jest stosunkowo rzadki i zwykle wtórny w stosunku do innych schorzeń lub terapii. Nie ma jednoznacznego punktu odcięcia dla stężenia triglicerydów, ale za wartość graniczną świadczącą o hipertriglicerydemii można przyjąć stężenie <35 mg/dL. Przyczynami mogą być rzadkie choroby genetyczne (abetalipoproteinemia, hipobetalipoproteinemia) oraz niedożywienie (w przypadku głodzenia czy zaburzonego wchłaniania np. w chorobie trzewnej).
Hirsutyzm to stan, w którym u kobiet występuje nadmierne owłosienie. Przypadłość ta dotyczy szczególnie obszarów ciała, które u większości kobiet są mniej owłosione, przede wszystkim twarzy (np. górna warga i broda), klatki piersiowej, pleców, brzucha, pośladków czy wewnętrznej strony ud. Chociaż umiarkowane owłosienie jest u kobiet naturalne, nadmierny wzrost włosów w obszarach typowych dla mężczyzn może wpływać na samopoczucie i pewność siebie pacjentek. Kiedy owłosienie można uznać za nadmierne? I jakie badania należy wykonać w trakcie diagnostyki hirsutyzmu?
Skąd bierze się hirsutyzm – nadmierne owłosienie u kobiet?
Hirsutyzm może wynikać ze zbyt dużego wpływu męskich hormonów płciowych, zwanych androgenami, lub być rezultatem stosowania leków o właściwościach androgennych.
Objawy związane z nadmiernym wydzielaniem męskich hormonów płciowych, czyli zespołem hiperandrogenizacji, mogą pojawić się w wyniku różnych czynników. Oto kilka z nich:
Choroby jajników: u niektórych kobiet hirsutyzm jest związany z chorobami jajników, np. z zespołem policystycznych jajników (PCOS). Osoby dotknięte PCOS mogą doświadczać nadmiernego owłosienia, nadwagi, niepłodności oraz problemów skórnych, takich jak trądzik. Guzy jajnika, które produkują androgeny, również mogą wywoływać te objawy.
Choroby nadnerczy: nadmierny wzrost owłosienia mogą powodować choroby nadnerczy, takie jak guzy wydzielające androgeny, wrodzony przerost nadnerczy i zespół Cushinga.
Podwyższone stężenie prolaktyny (hiperprolaktynemia): niektóre stany chorobowe, np. guzy produkujące prolaktynę, oraz przewlekły stres i długotrwały wysiłek fizyczny mogą powodować nadmierny wzrost owłosienia
Leki: hirsutyzm może być wynikiem stosowania pewnych leków, w tym danazolu, androgenów, steroidów anabolicznych i doustnych środków antykoncepcyjnych zawierających androgenny progestagen.
Nieznane przyczyny: w niektórych przypadkach nie udaje się ustalić jednoznacznego czynnika, który wywołuje hirsutyzm.
Jeśli u nastolatki występuje nadmierny wzrost owłosienia, często jest to zjawisko naturalne, związane z procesem dojrzewania. W takich przypadkach nie ma zazwyczaj powodów do niepokoju związanego z chorobami nowotworowymi. Niemniej jednak dla lekarza istotne jest, aby dokładnie ustalić, kiedy objawy nadmiernego owłosienia się pojawiły i jak szybko się nasilały.
Jeśli nadmierny wzrost owłosienia pojawił się nagle i rozwija się szybko, lekarz powinien wykluczyć obecność guzów jajników lub nadnerczy. W takich przypadkach konieczne jest szybkie działanie diagnostyczne. Dopiero po wykluczeniu poważniejszych onkologicznych przyczyn nadmiernego owłosienia można skupić się na diagnozowaniu innych, mniej groźnych schorzeń.
Jak często występuje hirsutyzm?
Hirsutyzm występuje stosunkowo często i może dotyczyć różnych grup wiekowych kobiet. Szacuje się, że pojawia się u 5–15% kobiet w wieku rozrodczym. Wskaźnik ten może się jednak różnić w zależności od populacji i czynników genetycznych.
Diagnostyka hirsutyzmu
Proces diagnostyczny jest niezbędny, aby lekarze mogli poznać przyczynę problemu, dokładnie go ocenić i zalecić właściwe działania. Oto główne aspekty diagnostyki hirsutyzmu:
Wywiad medyczny i historia pacjenta
Podczas pierwszej wizyty lekarz endokrynolog przeprowadza wywiad medyczny, aby uzyskać dokładne informacje na temat objawów hirsutyzmu, w tym:
Rodzaju i lokalizacji owłosienia: rozpoznawanie hirsutyzmu polega na analizie nasilenia nadmiernego owłosienia, a jednym z narzędzi, które pomagają w tym procesie, jest skala Ferrimana i Gallweya. Bazuje ona na ocenie rodzaju i rozmieszczenia włosów w dziewięciu obszarach ciała, które są szczególnie wrażliwe na działanie męskich hormonów płciowych: wardze górnej, brodzie, klatce piersiowej, brzuchu, podbrzuszu, plecach (górna i dolna część), pośladkach oraz wewnętrznych powierzchniach ud. Wyniki oceny opartej na tej skali pomagają określić stopień nadmiaru androgenów u pacjentki.
Czasu trwania objawów: pacjentka zostanie zapytana o to, kiedy zaobserwowała nadmierny wzrost owłosienia i czy objawy uległy pogorszeniu.
Historii rodzinnej: lekarz może zapytać o przypadki hirsutyzmu w rodzinie, co sugerowałoby czynnik genetyczny.
Historii medycznej: ważne jest, aby udzielić lekarzowi informacji na temat chorób, w tym zaburzeń hormonalnych, takich jak zespół policystycznych jajników (PCOS), a także przyjmowanych leków i istotnych schorzeń.
Badania fizykalne
Lekarz dokładnie bada pacjentkę, oceniając stan skóry i włosów na obszarach dotkniętych hirsutyzmem. Ponadto przeprowadza wywiad i badania fizykalne, które mają na celu zidentyfikowanie innych niż nadmierne owłosienie objawów hirustyzmu, takich jak zmiany skórne (trądzik, łojotok), nieregularne cykle menstruacyjne, problemy z płodnością, nadwaga lub otyłość, zmiany emocjonalne (depresja, lęki).
Badania laboratoryjne
Aby dokładnie zdiagnozować przyczynę hirsutyzmu, lekarz może zlecić badania laboratoryjne, w tym:
Badania hormonów: oznaczenie poziomu hormonów, takich jak: testosteron, DHEA-S (siarczan dehydroepiandrostendionu), prolaktyna. Pomocne mogą być również badania poziomów17-OH progesteronu, DHEA(dehydroepiandrosteron), DHT(dihydrotestosteron)czy hormonów tarczycy. Pozwoli to na wykluczenie lub potwierdzenie zaburzeń hormonalnych.
Badania metaboliczne: pacjentka może być skierowana na badania poziomu glukozy we krwi i insuliny, ponieważ insulinooporność jest często związana z PCOS.
Badania obrazowe: w niektórych przypadkach, aby dokładniej ocenić narządy wewnętrzne, takie jak jajniki czy nadnercza, może być zlecone przeprowadzenie badań obrazowych, np. ultrasonografii (USG).
Jak radzić sobie z hirsutyzmem – nadmiernym owłosieniem?
Metoda na walkę z hirsutyzmem zależna jest od jego przyczyny. Istnieje wiele sposobów terapii:
Leczenie farmakologiczne: lekarz może przepisać leki, które pomogą w regulacji poziomu hormonów i zmniejszeniu nadmiernego owłosienia.
Odstawienie leków wywołujących hirsutyzm: w przypadku hirsutyzmu spowodowanego lekami konieczne może być odstawienie medykamentu, który powoduje nieprawidłowy wzrost owłosienia.
Zabiegi kosmetyczne: zabiegi kosmetyczne, takie jak depilacja, pozwalają szybko pozbyć się owłosienia. Do metod tych zalicza się depilację laserową, która jest skuteczna w redukcji owłosienia, oraz mniej trwałe sposoby, np. depilację woskiem.
Zmiana stylu życia: jeśli hirsutyzm jest związany z chorobą, np. z PCOS, zmiana diety i regularna aktywność fizyczna mogą pomóc w kontrolowaniu objawów.
Chirurgiczne usunięcie guzów: jeśli hirsutyzm wynika z obecności guzów jajnika lub nadnerczy, kluczową procedurą jest ich chirurgiczne usunięcie.
Wsparcie psychologiczne: hirsutyzm wpływa na stan emocjonalny pacjentek. Terapia psychologiczna może pomóc w radzeniu sobie z negatywnymi emocjami i poprawić poczucie własnej wartości.
Niezależnie od przyczyny i stopnia nasilenia hirsutyzmu istnieją skuteczne sposoby leczenia tego schorzenia. Warto również stosować metody ukierunkowane na poprawę samooceny. Należy jednak podkreślić, że skuteczność zabiegów kosmetycznych może być ograniczona, jeśli nie zostanie ustalona przyczyna hirsutyzmu. Dlatego diagnoza jest kluczowym krokiem w wyborze odpowiedniego planu terapeutycznego, a leczenie powinno być dostosowane do konkretnej pacjentki.
Piśmiennictwo
E. Płaczkiewicz-Jankowska, Rozpoznawanie i leczenie hirsutyzmu u kobiet w wieku prokreacyjnym. Podsumowanie wytycznych Endocrine Society 2018, Medycyna Praktyczna 2018; 9: 34–39, https://www.mp.pl/endokrynologia/wytyczne/194403,rozpoznawanie-leczenie-hirsutyzmu-u-kobiet-w-wieku-prokreacyjnym
M. Góralska, U. Ambroziak, T. Bednarczuk, Hirsutyzm, zespół hiperandrogenizacji, https://www.mp.pl/pacjent/endokrynologia/choroby/168996,hirsutyzm-zespol-hiperandrogenizacji
P. Gajewski P., A. Szczeklik, Interna Szczeklika 2017, Medycyna Praktyczna, Kraków 2017, s. 1373–1375
A. Otlewska, P. Hackemer, F. Menzel, Hirsutyzm, Pediatria i Medycyna Rodzinna 2018; 14(4): 392–395
B. Żana, A. Jonas, Diagnostyka i leczenie hirsutyzmu u dziewcząt, Ginekologia Polska 2009; 80: 374–378
Miedź to bardzo ważny mikroelement w naszym organizmie. Jego zawartość w organizmie człowieka nie jest wysoka, jednak poprzez fakt, iż jest ona kofaktorem wielu reakcji enzymatycznych (dowiedz się więcej TUTAJ), jej rolę można zaobserwować w wielu miejscach naszego organizmu. Ostatnio coraz więcej mówi się o roli miedzi w chorobach tarczycy, niektórzy pacjenci chcieliby ją suplementować. Takie podejście nie jest uzasadnione, nadmierna suplementacja miedzi jest niekorzystna dla organizmu, zdecydowanie lepiej dostarczać ją z dietą i w ten sposób zapewnić optymalny poziom miedzi w organizmie.
Rola miedzi w chorobach tarczycy
Znaczenie miedzi w chorobach tarczycy ciągle jest kwestią badań. Na dziś wiemy, iż w badaniach na zwierzętach wykazano, iż podwyższony poziom miedzi związany jest z występowaniem niedoczynności tarczycy i podwyższonym poziomem TSH, natomiast stosunkowo niski poziom miedzi jest związany z nadczynnością tarczycy. Są również doniesienia wskazujące, iż poziom miedzi skorelowany jest z poziomem hormonów tarczycy. Redukcja stężenia miedzi może zwiększać stres oksydacyjny, powodując mniejszą produkcję hormonów tarczycy i spadek ich stężenia we krwi.
Badania eksperymentalne na myszach wskazują również, iż poziom hormonów tarczycy może wpływać na stężenie miedzi we krwi, poprzez zwiększenie jej uwalniania z magazynów w wątrobie. Badania przeprowadzone wśród pacjentów z nadczynnością tarczycy leczonych radioaktywnym jodem skutkowało zmniejszeniem poziomu hormonów tarczycy oraz poziomu miedzi.
Osobnym zagadnieniem jest korelacja pomiędzy stężeniem miedzi oraz chorobami autoimmunologicznymi tarczycy. Istnieją doniesienia, iż wysokie stężenie miedzi w surowicy jest skorelowane dodatnio z podwyższonym poziomem przeciwciał tarczycowych. Istnieją jednak doniesienia przeczące tej hipotezie, gdzie nie wykazano takiego powiązania.
Trwają również badania nad powiązaniem poziomu miedzi z nowotworami tarczycy. Uważa się, iż miedź rozpoczyna angiogenezę, czyli tworzenie naczyń krwionośnych w tkance nowotworowej, co sprzyja rozrostowi komórek złośliwych.
Podsumowując, doniesienia na temat wpływu miedzi na pracę tarczycy i produkcję przez nią hormonów wymagają dalszych badań. Wiemy, że miedź może mieć istotne znaczenie dla prawidłowej funkcji tego narządu. Biorąc jednak pod uwagę fakt, iż mamy do czynienia z mikropierwiastkiem, gdzie niewielkie wahania stężenia mają znaczenie, należy podkreślić, że dystrybucja i ilość biodostępnej miedzi musi być kontrolowana, co na dziś najłatwiej jest uzyskać poprzez spożycie miedzi z dietą.
Rola miedzi w chorobach układu sercowo-naczyniowego
Choroby układu sercowo-naczyniowego są jednymi z najczęstszych schorzeń występujących w krajach Europy Zachodniej i USA, są główną przyczyną śmiertelności. Miedź to pierwiastek śladowy, którego rola w patogenezie chorób serca nie była do tej pory rozpatrywana. Temat jednak ostatnio zyskuje na znaczeniu, rola miedzi jako kofaktora bardzo wielu reakcji enzymatycznych oraz czynnika biorącego udział w budowaniu kolagenu i elastyny skłania badaczy do przyjrzenia się temu zagadnieniu. Pojawiają się dowody wskazujące na to, iż brak równowagi w homeostazie miedzi przyczynia się do chorób serca. Zaburzenia regulacji białek transportujących miedź lub niedobory tego pierwiastka skutkują hipertofią mięśnia sercowego, niewydolnością serca, chorobą niedokrwienną serca czy kardiomiopatią cukrzycową.
Mechanizm wpływu miedzi na patogenezę chorób serca może być różny. Jednym z badanych zagadnień jest powiązanie stężenia ceruloplazminy – głównego białka transportującego miedź – z działaniem na czynność serca. Niedobór miedzi, zmniejszający aktywność ceruloplazminy i wpływający na homeostazę żelaza został powiązany z wystąpieniem cukrzycy, otyłości, dyslipidemii i miażdżycy. Coraz więcej badań klinicznych pokazuje, iż krążąca ceruloplazmina koreluje z ryzykiem chorób sercowo-naczyniowych oraz może być czynnikiem prognostycznym ryzyka tych chorób. Konieczne są jednak dalsze badania, aby zrozumieć, czy podwyższona krążąca ceruloplazmina odgrywa rolę w patogenezie chorób serca.
Innym mechanizmem, który jest rozpatrywany w powiązaniu poziomu miedzi i prawidłowej funkcji serca, jest jej udział w budowie kolageniu i elastyny. Nieprawidłowości w usieciowaniu tych białek skutkują mniejszą wytrzymałością tkanki łącznej, a co za tym idzie problemami w pracy serca, zaburzeniami skurczu i jego przerostem.
Choroby serca powodują ogromne obciążenia zdrowotne i ekonomiczne na całym świecie, i chociaż niedobór miedzi wpływa na wiele tkanek, w tym na wątrobę, jelita, układ naczyniowy, mózg i tkankę tłuszczową, serce wydaje się być jedną z najbardziej wrażliwych na niedobór miedzi tkanek.
Rola miedzi w chorobach układu nerwowego
Miedź odgrywa ważną rolę w funkcjonowaniu układu nerwowego poprzez swoje uczestnictwo w reakcjach enzymatycznych. Jej obecność w neuronach adrenergicznych warunkuje prawidłową aktywność β-hydroksylazy dopaminy (DBH), która przekształca dopaminę w noradrenalinę i jest niezbędna do równowagi katecholamin. W przypadku zaburzeń metabolizmu miedzi równowaga pomiędzy dopaminą a noradrenaliną zostaje zachwiana i wiąże się z wyższym poziomem dopaminy i niskim noradrenaliny w mózgu i w osoczu. Takie zmiany obserwowane są m.in. w genetycznie uwarunkowanej chorobie Menkesa (MNKD) i są wykorzystywane do jej diagnostyki.
W mózgu obecne są również pozostałe enzymy zależne od miedzi, jak ceruloplazmina, oksydaza lizylowa (sieciowanie kolagenu), SOD3, jednak ich rola w OUN nie została jeszcze wyjaśniona.
Oprócz roli kofaktora enzymów, miedź bierze udział w wielu procesach komórkowych, w których pełni rolę regulacyjną i sygnalizacyjną. Jest niezbędna do mielinizacji neuronów, moduluje funkcje receptorów GABA i NMDA, dzięki czemu wpływa na przepływ neurotransmiterów w synapsach. Dlatego przy niedoborze miedzi w diecie obserwuje się zaburzenia metaboliczne, zaburzenia mielinizacji neuronów, drgawki. Szkodliwy jest również nadmiar miedzi. Na przykład w chorobie Wilsona – genetycznie uwarunkowanej chorobie spichrzeniowej – miedź gromadzi się w mózgu i innych tkankach, a wzrost jej poziomu powoduje szerokie spektrum patologii neurologicznych i psychiatrycznych, w tym depresję, epizody psychotyczne, dystonię, drżenie i zaburzenia snu. Zaburzenie równowagi miedzi opisano także w przypadku choroby Parkinsona i Alzheimera, mogą może być również czynnikiem przyczyniającym się do etiologii innych zaburzeń neurodegeneracyjnych i starzenia się.
Wpływ miedzi na płodność u mężczyzn
Miedź odgrywa ważną rolę w męskiej płodności, jest elementem niezbędnym do produkcji męskich gamet oraz w procesach podziału komórek, takich jak mitoza i mejoza.
Enzymy, w których miedź jest kofaktorem (ceruloplazmina, dysmutaza nadtlenkowa SOD1 i SOD2, oksydaza cytochromu C), są również obecne na wszystkich etapach gametogenezy, a ponadto w komórkach somatycznych jąder i najądrzy oraz w płynach związanych z plemnikami w prostacie i najądrzach.
Opisano również udział miedzi w dystrybucji androgenów – męskich hormonów płciowych – na osi podwzgórze – przysadka – jądro.
Stąd zarówno wzrost stężenia miedzi, jak i jej niedobór, prowadzi do zaburzeń płodności. Wpływ miedzi na te procesy nabiera większego znaczenia wobec faktu, iż problem niepłodności dotyczy coraz większej liczby osób, a środowisko jest coraz bardziej zanieczyszczone.
Diagnostyka zawartości miedzi w organizmie człowieka – testy z krwi i z moczu, kiedy zrobić badanie?
Miedź to pierwiastek niezbędny do prawidłowego rozwoju organizmów żywych, poprzez uczestnictwo w bardzo wielu reakcjach enzymatycznych. Dlatego niedobór miedzi w sposób istotny wpływa na redukcję lub całkowite zablokowanie enzymów zależnych od miedzi, hamując w ten sposób niektóre procesy życiowe. Z drugiej strony miedź jako pierwiastek reaktywny może powodować powstawanie wolnych rodników, co w konsekwencji może doprowadzić do uszkodzenia DNA oraz białek. Dlatego organizm człowieka wypracował precyzyjne mechanizmy regulujące stężenie miedzi w komórkach. Przyjmowanie suplementów miedzi i cynku może zaburzać tę równowagę. Miedź powinna być dostarczana z pożywienia.
Stany wymagające ewentualnej suplementacji miedzią obejmują:
zaburzenia wchłaniania związane z celiakią i chorobami trzustki,
choroby nerek,
wykryte niedobory miedzi.
Badanie poziomu miedzi w organizmie można wykonać z krwi i z moczu.
Wskazania do wykonania poziomu miedzi w surowicy obejmują:
podejrzenie choroby Menkesa,
podejrzenie choroby Wilsona – zaburzenia wbudowywania miedzi do ceruloplazminy,
marskość wątroby,
podejrzenie niedoborów lub nadmiaru miedzi w organizmie.
Badanie poziomu miedzi w moczu jest badaniem przydatnym w diagnostyce i monitorowaniu choroby Wilsona. Jego przydatność w oznaczaniu poziomu miedzi w organizmie jest ograniczona.
Piśmiennictwo
Zhou Q, Xue S, Zhang L, Chen G. Trace elements and the thyroid. Front Endocrinol (Lausanne). 2022 Oct 24;13:904889. doi: 10.3389/fendo.2022.904889. PMID: 36353227; PMCID: PMC9637662.
Liu Y, Miao J. An Emerging Role of Defective Copper Metabolism in Heart Disease. Nutrients. 2022 Feb 7;14(3):700. doi: 10.3390/nu14030700. PMID: 35277059; PMCID: PMC8838622.
Lutsenko S, Washington-Hughes C, Ralle M, Schmidt K. Copper and the brain noradrenergic system. J Biol Inorg Chem. 2019 Dec;24(8):1179-1188. doi: 10.1007/s00775-019-01737-3. Epub 2019 Nov 5. PMID: 31691104; PMCID: PMC6941745.
Ogórek M, Gąsior Ł, Pierzchała O, Daszkiewicz R, Lenartowicz M. Role of copper in the process of spermatogenesis. Postepy Hig Med Dosw (Online). 2017 Aug 9;71(0):663-683. doi: 10.5604/01.3001.0010.3846. PMID: 28791960.
Liczba pojęć pojawiająca się na wyniku naszego profilu lipidowego może przyprawić o zawroty głowy: cholesterol, trójglicerydy, lipoproteiny, frakcje cholesterolu, frakcje lipoprotein, gęstości lipoprotein. Zrozumienie dodatkowo utrudniają wartości poszczególnych parametrów lipidogramu oraz ich zakresy referencyjne. Biorąc pod uwagę, że zwykle profil lipidowy nie jest jedynym badaniem, które omawiamy z lekarzem w trakcie wizyty w gabinecie, często brakuje czasu na dokładne wytłumaczenie uzyskanych wyników oraz tego, co oznaczają poszczególne wartości. Z tego artykułu dowiesz się, jak działa gospodarka lipidowa w naszym organizmie i co oznaczają poszczególne parametry lipidogramu/profilu lipidowego.
Lipidy – cholesterol, trójglicerydy
Lipidy są szeroką grupą związków organicznych, które charakteryzuje nierozpuszczalność w wodzie. W kontekście profilu lipidowego mówimy o dwóch grupach lipidów – cholesterolu i trójglicerydach.
Cholesterol
Cholesterol jest złożoną cząsteczką o charakterystycznym szkielecie składającym się z połączenia czterech pierścieni zbudowanych z 17 atomów węgla. Ta specyficzna budowa jest wspólna dla steroidów, czyli grupy, do której należą również witamina D, kwasy żółciowe czy też hormony steroidowe: płciowe (np. testosteron, estrogeny czy progesteron), mineralokortykoidy (np. aldosteron odpowiadający za gospodarkę wodno-elektrolitową) czy glikokortykosteroidy (np. kortyzol popularnie nazywany hormonem stresu).
Cholesterol pełni wiele ważnych funkcji w naszym organizmie:
Wspieranie integralności błony komórkowej: jest kluczowy dla utrzymania płynności i integralności strukturalnej błon komórkowych. Reguluje przepuszczalność błony i wspiera funkcję białek błonowych pełniących różnorodne funkcje (np. receptorową, transportową);
Synteza hormonów steroidowych: cholesterol służy jako cząsteczka prekursorowa dla syntezy wspomnianych wcześniej hormonów steroidowych.
Synteza witaminy D: cholesterol jest również prekursorem witaminy D, która jest syntetyzowana, gdy skóra jest narażona na działanie światła ultrafioletowego. Witamina D odgrywa kluczową rolę w metabolizmie wapnia i zdrowiu kości.
Synteza kwasów żółciowych: cholesterol jest przekształcany w kwasy żółciowe w wątrobie, które następnie są magazynowane w pęcherzyku żółciowym jako składnik żółci, która jest niezbędna do emulgowania i trawienia tłuszczów przyjmowanych z pokarmem.
Wsparcie funkcjonowania układu nerwowego: cholesterol jest istotny dla mielinizacji komórek nerwowych, co sprzyja efektywnemu przewodnictwu sygnałów między komórkami nerwowymi.
Należy zaznaczyć, że pomimo iż cholesterol jest niezbędny dla wyżej wskazanych funkcji, organizm potrzebuje bardzo małych jego ilości do utrzymania tych procesów w pełnej sprawności. Stąd, wbrew powszechnym obawom, obniżanie stężenia cholesterolu nie doprowadzi do upośledzenia tych szlaków metabolicznych i pogorszenia stanu zdrowia.
Człowiek pobiera około 70-80% cholesterolu z pożywienia, a 20-30% jest syntetyzowane przez człowieka w wątrobie oraz jelicie cienkim. Co ciekawe, cholesterol nie przekracza bariery krew-mózg, dlatego jest on lokalnie syntetyzowany na potrzeby ośrodkowego układu nerwowego.
Trójglicerydy
Triglicerydy lub inaczej trójglicerydy (potocznie zwane tłuszczami) – są to cząsteczki składające się z połączenia glicerolu oraz trzech łańcuchów kwasów tłuszczowych (o długości od 14 do 24 atomów węgla oraz różnej liczbie podwójnych wiązań między nimi). Łańcuchy te mogą być zarówno te same we wszystkich trzech pozycjach, jak i różne – co wpływa na ich biologiczne właściwości. W zależności od położenia łańcuchów w cząsteczce zmieniają się właściwości biologiczne cząsteczki.
Trójglicerydy pełnią następujące funkcje w organizmie:
Źródło energii: trójglicerydy dostarczają dużo więcej energii w porównaniu do węglowodanów i białka. Glicerol i kwasy tłuszczowe uwalniane z rozkładu kwasów tłuszczowych mogą zostać wykorzystane przez komórkę do produkcji energii.
Przechowywanie energii: trójglicerydy sąmagazynowane w tkance tłuszczowej i mogą stanowić dodatkowe źródło do wyprodukowania energii przez komórki.
Transport kwasów tłuszczowych: kwasy tłuszczowe mogą zostać wykorzystane także do produkcji licznych istotnych biologicznie pochodnych. 99% wszystkich kwasów tłuszczowych we krwi jest transportowane w połączeniu z glicerolem.
Lipoproteiny – uniwersalny nośnik lipidów
Jako wspomniano wcześniej wspólną cechą zarówno trójglicerydów, jak i cholesterolu jest nierozpuszczalność w wodzie. Hipotetycznie, obie frakcje dostając się do osocza krwi w wolnej postaci, zachowałyby się jak olej wlany do wody. Dlatego, dla efektywnego transportu w osoczu krwi, lipidy posiadają swój przenośnik – lipoproteiny.
Lipoproteiny są dużymi strukturami, które, jak podpowiada nam nazwa, składają się lipidów i białek. Ich struktura jest jednak bardziej złożona, co wyjaśnia poniższa rycina. Cząsteczkę lipoproteiny można porównać do bańki. Ściany tej bańki składają się z fosfolipidów – trzeciej, innej grupy lipidów posiadającej szczególną właściwość posiadania hydrofilowej „głowy” oraz hydrofobowego „ogona”, co umożliwia stworzenie bariery między wodą, a nierozpuszczalnymi w niej tłuszczami. W ten sposób lipoproteina może mieszać się z osoczem krwi i jednocześnie efektywnie utrzymywać lipidy w swoim wnętrzu. Na powierzchni, w „ścianie bańki” znajdują się zatopione białka m.in. apolipoproteiny (apo z gr. ἀπό od/z dala od – w tym kontekście oddzielające się od lipoprotein) kluczowe dla przenoszenia lipidów między lipoproteinami a komórkami organizmu.
Lipoproteiny nie są jednolitą grupą i poszczególne typy wykazują różne biologiczne funkcje w organizmie. Historycznie, na podstawie rozdziału za pomocą wirowania (ultracentryfugacji) i ruchomości w polu elektrycznym (elektroforezy), lipoproteiny podzielono na następujące frakcje:
Chylomikrony – zawierają głównie trójglicerydy (~90%) i odpowiadają za transport lipidów z przewodu pokarmowego do wątroby drogami limfatycznymi. Przeważnie poza okresem po posiłku nie pojawiają się we krwi obwodowej.
Lipoproteiny o bardzo małej gęstości (z ang. Very-Low-Density Lipoproteins – VLDL) – składające się w 45-50% z trójglicerydów i 30% z cholesterolu. Ich główną funkcją jest transport lipidów z wątroby do innych tkanek.
Lipoproteiny o średniej gęstości (z ang. Intermediate-Density Lipoproteins – IDL) – powstają z oddawania przez VLDL trójglicerydów do tkanek obwodowych, swoiście „zagęszczając się” – zmniejsza się zawartość trójglicerydów, a zwiększa cholesterolu.
Lipoproteiny o małej gęstości (z ang. Low-Density Lipoproteins – LDL) oraz lipoproteina(a) – LDL mogą powstawać w wyniku dalszego „zagęszczania” IDL lub mogą być bezpośrednio wydzielane przez wątrobę. Do LDL należy m.in. cholesterol, który stanowi ~70% wszystkich lipidów wewnątrz tej grupy. Lipoproteina(a) jest podobnie w budowie i składzie do LDL, jednak wyróżnia się dodatkową cząsteczką apolipoproteiny(a) na jej powierzchni.
Lipoproteiny o dużej gęstości (z ang. High-Density Lipoproteins – HDL) – składają w znacznej mierze z białek. Cholesterol stanowi ~40%, a trójglicerydy ~5% lipidów wewnątrz HDL. Główną rolą HDL jest wsteczny transport cholesterolu z tkanek obwodowych do wątroby.
Ze zwiększającą się gęstością maleje wielkość lipoprotein (HDL może być nawet 15 razy mniejszy od dużej cząsteczki VLDL). Mniejsze cząsteczki są również dużo liczniejsze od tych większych: średnio u zdrowej osoby na jeden chylomikron przypadnie około 200 cząsteczek VLDL, 1500 cząsteczek IDL, 2000 cząsteczek LDL i kilkanaście tysięcy cząsteczek HDL. Z tego też względu większość cholesterolu (~70%) jest zamknięte w LDL i HDL, natomiast VLDL i IDL transportują około 80% wszystkich trójglicerydów. Dlatego LDL nazywamy lipoproteinami bogatymi w cholesterol, a IDL and VLDL lipoproteinami bogatymi w trójglicerydy.
Charakterystyczną cechą chylomikronów, VLDL, IDL i LDL jest występowanie na ich powierzchni apolipoproteiny B. Z kolei na powierzchni HDL występuje apolipoproteina A1.
Lipoproteiny a rozwój miażdżycy
Rozwój miażdżycy jest skomplikowanym, wieloetapowym procesem, w którym kluczową rolę odgrywają lipoproteiny bogate w cholesterol (głównie LDL). W sytuacji nadmiernego stężenia tych lipoprotein we krwi, zwiększa się ich przenikanie do ściany tętnicy. Po dostaniu się do ściany naczyniowej, te lipoproteiny oddają cholesterol makrofagom. Makrofagi te, akumulując cholesterol, przekształcają się w tzw. komórki piankowate, które są głównymi elementami tworzącymi blaszkę miażdżycową.
Ostatnie badania naukowe wskazują na to, że to niekoniecznie sama zawartość lipidów w tych lipoproteinach jest problemem, ale raczej ich liczba – nośnik, a nie zawartość. Okazuje się, że VLDL i IDL jako nośniki cholesterolu również mogą uczestniczyć w powstawaniu miażdżycy. Z tego względu kluczowe w ograniczaniu miażdżycy jest zmniejszanie liczby wszystkich lipoprotein zawierających apolipoproteinę B – a więc VLDL, IDL i LDL.
W odniesieniu do lipoprotein HDL, które tradycyjnie były uznawane za nośnik „dobrego cholesterolu” ze względu na ich zdolność do usuwania cholesterolu z krwi i tkanek, najnowsze badania wprowadzają pewne wątpliwości co do ich roli. Okazuje się, że niskie stężenie HDL może nie być przyczyną problemów, ale raczej skutkiem zwiększonej liczby lipoprotein VLDL, IDL i LDL. To odkrycie sugeruje, że obniżenie poziomu HDL może być efektem ubocznym zwiększonego stężenia innych, bardziej aterogennych lipoprotein, a nie wynikiem utraty niezależnego mechanizmu ochronnego.
Profil lipidowy – spojrzenie na lipoproteiny przez dziurkę od klucza
Pomimo, że to liczba cząsteczek lipoprotein VLDL, IDL, LDL, a nie stężenie poszczególnych lipidów jest bezpośrednio związane z rozwojem miażdżycy, oznaczenie stężeń może być prostym i użytecznym sposobem przybliżenia liczby cząsteczek lipoprotein.
W profilu lipidowym znajdujemy następujące parametry:
stężenie trójglicerydów – biorąc pod uwagę, że większość trójglicerydów jest przenoszona przez VLDL i IDL, ich stężenie będzie przybliżać liczbę cząsteczek lipoprotein VLDL i IDL – bogatych w trójglicerydy. Względna zawartość (zagęszczenie) trójglicerydów w tych cząsteczkach jest zmienna, stąd też przybliżenie to nie jest idealne.
stężenie cholesterolu HDL – z racji, że cholesterol jest głównym lipidem w HDL, jego stężenie może przybliżać liczbę cząsteczek HDL. Nie jest to, podobnie jak w przypadku trójglicerydów, przybliżenie doskonałe, ale wystarczające w praktyce klinicznej.
stężenie cholesterolu LDL – względna zawartość cholesterolu w cząsteczkach LDL jest prawie stała, dlatego też stężenie cholesterolu w LDL będzie bezpośrednim wskaźnikiem liczby cząsteczek LDL, w ten sposób określając rzeczywiste ryzyko sercowo-naczyniowe związane z LDL.
stężenie cholesterolu nie-HDL – różnica całkowitego cholesterolu i cholesterolu HDL jest równa całkowitej zawartości cholesterolu w lipoproteinach aterogennych (VLDL, IDL, LDL). Okazuje się, że cholesterol w IDL i LDL może być dobrym wskaźnikiem liczby tych cząsteczek. Stąd jego suma z cholesterolem LDL może odpowiadać całkowitemu potencjałowi promiażdzycowemu związanego z lipidami.
stężenie cholesterolu całkowitego – będzie to wypadkowa suma cholesterolu we wszystkich frakcjach lipoprotein. Przy braku zwiększonej liczby VLDL i IDL (na przykład w otyłości, cukrzycy, zespole metabolicznym) wzrost całkowitego stężenia cholesterolu będzie wynikał głównie ze wzrostu cholesterolu LDL i spadku cholesterolu HDL. Jednak biorąc pod uwagę złożoność tego parametru (zmienność HDL) cholesterol LDL i nie-HDL są lepszymi wskaźnikami ryzyka sercowo-naczyniowego.
Dodatkowymi parametrami profilu lipidowego może być:
Apolipoproteina B – biorąc pod uwagę, że każda cząsteczka VLDL, IDL i LDL ma na swojej powierzchni jedną cząsteczkę apolipoproteiny B, jej stężenie będzie odzwierciedlało całkowite stężenie VLDL, IDL i LDL – a więc całkowity potencjał promiażdzycowy związanego z lipidami.
Apolipoproteina A1 – podobnie do sposobu, w jaki apolipoproteina B przybliża liczbę wszystkich VLDL, IDL i LDL, apolipoproteina A1 może przybliżać bezpośrednio liczbę lipoprotein HDL.
Lipoprotein(a) – jej poziom jest determinowany w dużej mierze genetycznie. Z racji podobieństwa do LDL, w przypadku dużego stężenia lipoproteina(a) podwyższone stężenia cholesterolu całkowitego, cholesterolu nie-HDL mogą być zafałszowane cholesterolem przenoszonym w lipoproteinie(a), a nie bezpośrednio w zwykłym LDL. Ma to znaczenie terapeutyczne, ponieważ klasyczne leki obniżające stężenie lipidów nie wpływają istotnie na stężenie lipoproteiny(a), dlatego może to prowadzić do sytuacji, że terapia nie będzie prowadzić do obniżenia stężenia cholesterolu nie-HDL, czyli: liczby cząsteczek VLDL, IDL i LDL, gdyż rzeczywista przyczyna będzie leżała w lipoproteinie(a).
Piśmiennictwo
Visseren FLJ, Mach F, Smulders YM, et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). European Heart Journal. 2021;42(34):3227-3337.
Banach M, Burchardt P, Chlebus K, et al. Wytyczne PTL/KLRWP/PTK/PTDL/PTD/PTNT diagnostyki i leczenia zaburzeń lipidowych w Polsce 2021. Nadciśnienie Tętnicze w Praktyce. 2021;7(3):113-122.
Stłuszczenie wątroby to stan, w którym w hepatocytach (komórkach wątroby) gromadzi się tłuszcz. Więcej o stłuszczeniu wątroby przeczytacie TUTAJ, ten artykuł będzie poświęcony jednej z postaci stłuszczenia wątroby, dawniej nazywaną niealkoholowym stłuszczeniem wątroby (NAFLD), a obecnie stłuszczeniową chorobą wątroby związaną z zaburzeniami metabolicznymi (MASLD). Dlaczego zmieniono nazwę choroby, jakie są jej objawy, jak się ją diagnozuje i leczy? Zapraszamy do przeczytania.
Stłuszczeniowa choroba wątroby związana z zaburzeniami metabolicznymi (MASLD)– dawniej niealkoholowe stłuszczenie wątroby (NAFLD)
Niealkoholowa stłuszczeniowa choroba wątroby – NAFLD (nonalcoholic fatty liver disease) jest najczęściej rozpoznawaną przewlekłą chorobą wątroby w krajach rozwiniętych, w bazie chorób ICD-10 sklasyfikowana jako K76 – Stłuszczenie wątroby niesklasyfikowane gdzie indziej. Obecnie nazwa choroby została zmieniona na stłuszczeniowa choroba wątroby związana z zaburzeniami metabolicznymi (MASLD). Badania laboratoryjne – oznaczanie glukuronidu etylu – u pacjentów z NAFLD wykazywały, iż część z nich spożywa jednak alkohol, dlatego nazwa niealkoholowe stłuszczenie wątroby nie była adekwatna. Ponadto nowa nazwa obejmuje pacjentów ze stłuszczeniem wątroby i co najmniej jednym z pięciu kardiometabolicznych czynników ryzyka.
Stłuszczeniowa choroba wątroby z zaburzeniami metabolicznymi definiowana jest jako stłuszczenie wątroby (widoczne w badaniu obrazowym lub histopatologicznym) u pacjenta, który nie spożywa więcej niż 20 g alkoholu dziennie – kobiety i 30 g – mężczyźni oraz spełnione jest przynajmniej jedno z poniższych kryteriów:
BMI ≥25 kg/m2 lub obwód talii ≥94 cm u mężczyzn i ≥80 cm u kobiet
ciśnienie tętnicze ≥130/85 mm Hg lub pacjent leczy się z powodu nadciśnienia tętniczego
poziom triglicerydów w surowicy ≥1,7 150 mg/dl lub pacjent leczy się z powodu hipertriglicerydemii
cholesterol HDL w surowicy ≤ 40 mg/dl u mężczyzn i ≤ (50 mg/dl u kobiet lub pacjent leczy się z powodu hipercholesterolemii
stężenie glukozy na czczo ≥ 100 mg/l lub 2 h po obciążeniu glukozą ≥140 mg/dl, lub HbA1c ≥5,7% lub cierpi/leczy się z powodu cukrzycy t.2.
Niska aktywność fizyczna, zwiększone spożycie kalorii powoduje zespół metaboliczny, insulinooporność, zwiększenie poziomu wolnych kwasów tłuszczowych (FFA) w hepatocytach, akumulację trójglicerydów (TG), stres oksydacyjny. Zwłóknienie wątroby nasila stan zapalny i powoduje śmierć komórek.
Schorzenie ma charakter postępujący, obserwuje się następujące etapy choroby:
stłuszczenie – prostego nacieku tłuszczowego,
MASH (metabolic dysfunction-associated steatohepatitis), dawniej NASH (non-alcoholic steatohepatitis uszkodzenie charakteryzujące się stanem zapalnym, zmianami zwyrodnieniowymi hepatocytów i marskością,
Do etapu marskości wątroby choroba często przebiega bezobjawowo i nie wpływa na samopoczucie chorego, dlatego najczęściej wykrywana jest przypadkowo, np. kiedy zostanie wykonane badanie USG wątroby albo w badaniu laboratoryjnym oznaczone aktywności enzymów wątrobowych (AsPAT, AlAT).
Jeśli objawy występują, to chorzy najczęściej skarżą się na:
Pacjenci z MASLD zwykle cierpią na otyłość, w badaniu przedmiotowym u ponad 75% osób można stwierdzić powiększoną wątrobę, a u ok. 25% – powiększoną śledzionę.
Konsekwencje i powikłania MASLD – niealkoholowej stłuszczeniowej choroby wątroby (NAFLD)
Pacjenci z prostym stłuszczeniem wątroby są „hepatologicznie” pacjentami bezpiecznymi. Natomiast obecność w wątrobie – obok stłuszczenia – także stanu zapalnego, w przebiegu którego dochodzi do uszkodzenia komórek wątrobowych, może powodować włóknienie wątroby. To stadium określa się mianem MASH (metabolic dysfunction-associated steatohepatitis), dawniej NASH (non-alcoholic steatohepatitis) i jest to etap pośredni między stłuszczeniem prostym a zaawansowanymi postaciami choroby. Stadium MASH/NASH rozpoznaje się na podstawie badania histologicznego, wykonanego z materiału pobranego w czasie biopsji wątroby.
Na szczęście progresywne warianty dotyczą zdecydowanej mniejszości pacjentów z tą chorobą – nie więcej niż 10%.
Proste stłuszczenie zazwyczaj uważa się za chorobę odwracalną, natomiast stadium MASH (dawniej NASH) uznaje się za przyczynę marskości wątroby. Wraz z rozwojem choroby stłuszczenie i zmiany zwyrodnieniowe hepatocytów zmniejszają się, rozwija się włóknienie narządu.
Włóknienie wątroby zazwyczaj postępuje powoli, u niektórych chorych zmiany pozostają stabilne przez wiele lat. U około 20% pacjentów postęp choroby jest jednak szybki, a przyczyny stabilności lub szybkiej dekompensacji nie są znane.
Marskość związana z niealkoholowym stłuszczeniowym zapaleniem wątroby (NASH) może rozwinąć się w raka wątrobowokomórkowego, a także nawracać po przeszczepie wątroby.
Procesy decydujące o progresji niealkoholowej stłuszczeniowej choroby wątroby (NAFLD)do marskości wątroby lub raka wątrobowokomórkowego nie są dobrze zbadane. Zalicza się do nich:
narastającą insulinooporność tkanek obwodowych i wątroby,
Dlaczego dochodzi do stłuszczenie wątroby, a następnie do jej włóknienia?
Insulinooporność jest cechą charakterystyczną zespołu metabolicznego. Głównym zaburzeniem w metabolizmie węglowodanów jest zmniejszenie wychwytu glukozy przez mięśnie (wzrost stężenia glukozy w surowicy), ale insulinooporność przyczynia się również do zwiększenia wychwytu kwasów tłuszczowych przez wątrobę, co powoduje zahamowanie cyklu Krebsa i stymulację glukoneogenezy, czego efektem jest zwiększone wytwarzanie glukozy w wątrobie. Taki stan hiperglikemii jest wykrywany przez komórki β- trzustki, które zwiększają produkcję insuliny w celu przywrócenia prawidłowej glikemii. Z czasem organizm traci zdolność do zapewnienia zwiększonej podaży insuliny i rozwija się cukrzyca. Natomiast głównym zaburzeniem metabolizmu lipidów jest oporność na stymulowane przez insulinę hamowanie lipolizy, co prowadzi do zwiększonego wytwarzania wolnych kwasów tłuszczowych z tkanki tłuszczowej. Kwasy tłuszczowe są dostarczane do wątroby, gdzie są ponownie estryfikowane do triglicerydów, dochodzi do ich kumulacji w hepatocytach (stłuszczenie!) oraz nasilonej β- oksydacji w mitochondriach, co z kolei powoduje powstawanie reaktywnych form tlenu (ROS) (w procesach tych uczestniczą też inne wątrobowe szlaki oksydacyjne np. cytochrom P-450).
W rozwoju stłuszczenia do MASH/NASH niezwykle istotne znaczenie ma również stres oksydacyjny. ROS powoduje hemotaksję komórek zapalnych, które uwalniają cytokiny i chemokiny prozapalne (np. TNF-α) co dodatkowo nasila stan zapalny w wątrobie. ROS i TNF-α bezpośrednio stymulują komórki gwiaździste wątroby do wytworzenia macierzy zewnątrzkomórkowej, co prowadzi do rozwoju włóknienia wątroby. ROS są odpowiedzialne za rozwój zaburzeń mitochondrialnych obserwowanych w NASH (peroksydacja lipidów – oksydacyjne uszkodzenie mitochondrialnego DNA). Niedobór przeciwutleniaczy np. witaminy E przyczynia się do uszkodzeń oksydacyjnych.
Diagnostyka stłuszczeniowej choroby wątroby (niealkoholowego stłuszczenia wątroby)
W rozpoznawaniu zaawansowanego włóknienia wątroby, które właściwie jest jedynym znanym czynnikiem o niekorzystnym rokowaniu, stosuje się dwuetapową diagnostykę włóknienia wątroby.
W pierwszym etapie wykorzystywany jest indeks FIB-4, oparty jest na czterech parametrach:
wieku pacjenta,
aktywności aminotransferaz – ALT i AST,
liczbie płytek krwi.
FIB-4 dzieli pacjentów ze stłuszczeniem wątroby na trzy grupy, określające prawdopodobieństwo zaawansowanego włóknienia wątroby. Chorzy o wysokim i średnim ryzyku mają wykonywane badanie elastograficzne, oceniające sztywność wątroby (koreluje ona z ilością tkanki łącznej w tym narządzie). Dopiero w przypadku niejednoznacznych wyników badań nieinwazyjnych wykonuje się biopsję wątroby.
Badacze wykazali, że wykorzystując wskaźnik FIB-4 można prawidłowo wytypować pacjentów do biopsji wątroby i jednocześnie wskazać tych, którzy biopsji mogą uniknąć.
Leczenie niealkoholowej stłuszczeniowej choroby wątroby (NAFLD)
1. Podstawowe znaczenie w leczeniu choroby ma zmiana stylu życia – odpowiednia dieta i aktywność fizyczna.
Dieta – u pacjentów z nadwagą i otyłością celem jest zmniejszenie masy ciała o co najmniej 10%, co przekłada się na zmniejszenie ilości tłuszczu w wątrobie, zmniejszenie procesu zapalnego i regresję włóknienia. Utrata 5% tłuszczu trzewnego przekłada się na redukcję tłuszczu w wątrobie o około 40%.
Wpływ właściwej diety i zerwanie z siedzącym trybem życia mają podstawowe znaczenie dla osiągnięcia redukcji masy ciała, która powinna być procesem długotrwałym z tygodniową redukcją na poziomie 0,5 kg. Przestrzega się pacjentów przed narzucaniem sobie zbyt dużych restrykcji dietetycznych, a zwłaszcza przed głodzeniem, które – poza problemami wynikającymi z niedoborów witaminowych – jest niebezpieczne dla wątroby. Zbyt szybki spadek masy zwiększa stłuszczenie wątroby, a w skrajnych przypadkach może nawet prowadzić do jej niewydolności.
U pacjentów z MASLD zaleca się wprowadzanie do jadłospisu elementów diety śródziemnomorskiej. Ta właśnie dieta, z jednoczesnym ograniczeniem węglowodanów, okazała się lepsza w aspekcie mobilizacji tłuszczu wątrobowego od diety niskotłuszczowej. Zaleca się również ograniczenie produktów słodzonych fruktozą oraz unikanie spożycia alkoholu, w przypadku występowania zwłóknienia od fazy drugiej wzwyż rekomendowana jest całkowita abstynencja.
Aktywność fizyczna – jej intensywność powinna być dostosowana do możliwości chorego, zajmować co najmniej 20–30 minut dziennie przez przynajmniej 5 dni w tygodniu, w zależności od jego intensywności. Rekomendacje mówią, iż powinno to być 150-300 minut wysiłku o umiarkowanej intensywności tygodniowo, lub 75-150 minut wysiłku o dużej intensywności. Redukcja ilości tłuszczu w wątrobie jest proporcjonalna do zwiększenia aktywności fizycznej.
2. Leczenie farmakologiczne znajduje zastosowanie u chorych z MASH potwierdzonym przez badanie histologiczne. Stosuje się m.in. witaminę E, chociaż jej stosowanie ma pewne ograniczenia i należy przestrzegać zaleceń lekarza. Pacjent z MASLD powinien być objęty leczeniem chorób współtowarzyszących – otyłości, nadciśnienia, hiperlipidemii.
3. Przeszczep wątroby jest leczeniem stosowanym u pacjentów ze schyłkową marskością wątroby lub rakiem wątrobowokomórkowym.
Podsumowanie
Dotychczas najczęstszą przyczyną marskości wątroby była infekcja HCV. Ale obecnie, dzięki dostępowi do skutecznych leków oraz programom przesiewowym, zdekompensowana marskość wątroby o etiologii wirusowej jest coraz rzadszym zjawiskiem, jej miejsce natomiast zajmuje marskość na podłożu stłuszczeniowej choroby wątroby związanej z zaburzeniami metabolicznymi MASLD. Stłuszczenie metaboliczne jest nie tylko najczęstszą przewlekłą chorobą wątroby, ale staje się też najczęstszą przyczyną transplantacji wątroby.
Piśmiennictwo
MG Scott, AM Gronowski, CHS Eby: Tietz Medycyna laboratoryjna w praktyce- przypadki kliniczne, Medpharm Polska, Wrocław 2016.
A Brzozowska, M Olszanecka-Glinianowicz: Pacjent z NAFLD- rola dietetyka oraz jego współpraca z lekarzem, Medycyna po dyplomie wrzesień 2020.
Prof.M.Hartleb: Co to jest niealkoholowa stłuszczeniowa choroba wątroby?-wywiad, Medycyna praktyczna.
Sterling RK, Lissen E, Clumeck N, Sola R, Correa MC, Montaner J, S Sulkowski M, Torriani FJ, Dieterich DT, Thomas DL, Messinger D, Nelson M; APRICOT Clinical Investigators.
Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006 Jun
Mach T., Szczepanek M, Stłuszczeniowa choroba wątroby związana z zaburzeniami metabolicznymi, https://www.mp.pl/interna/chapter/B16.II.7.11. Dostęp 08.02.2025
Neubauer K., MASLD. Zasady rozpoznawania i leczenia. Wykład na konferencji IV Ogólnopolska Konferencja Kardiologia i Choroby Metaboliczne Wieku Podeszłego. 8-9.01.2025, dostęp online.
Lamblioza (inaczej giardioza) jest najbardziej powszechną na świecie pierwotniakową chorobą dwunastnicy i jelita cienkiego przebiegającą pod postacią biegunki. Szacuje się, że zarażenie pasożytem Giardia lamblia dotyczy w naszym kraju ok. 10% populacji dorosłych i nawet do 25-50% populacji dzieci. Rocznie odnotowuje się w Polsce ponad 2 tys. objawowych przypadków zarażenia. Przyswojenie od najmłodszych lat prawidłowych nawyków higienicznych może w znacznym stopniu zredukować prawdopodobieństwo zarażenia.
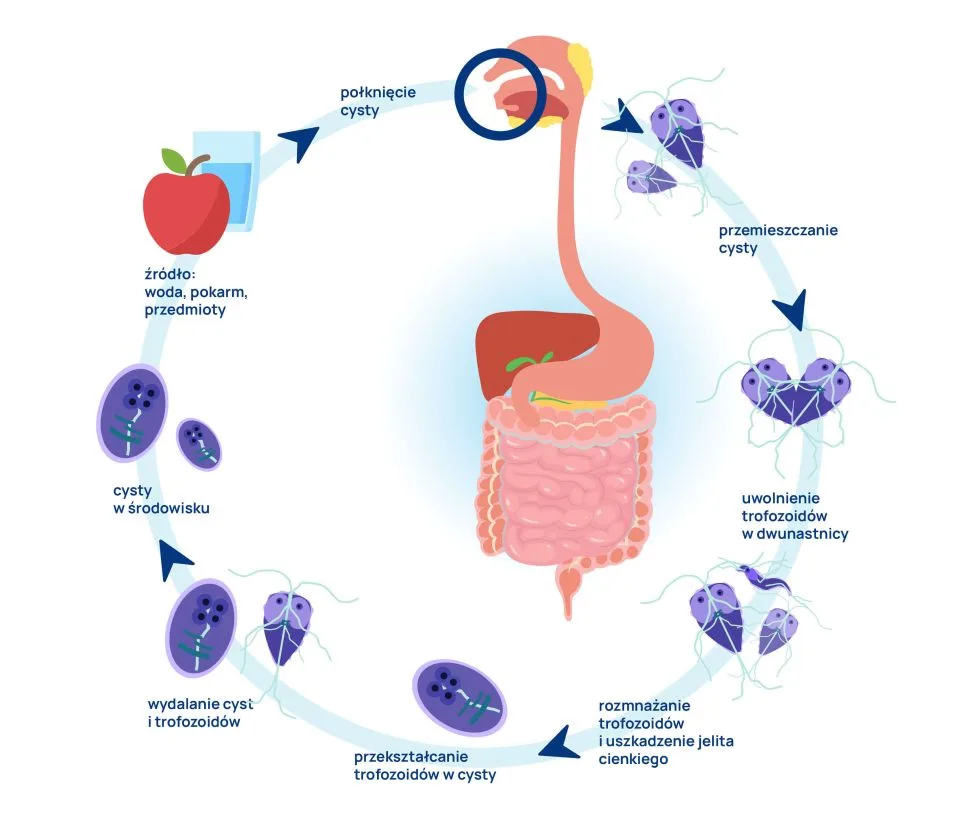
Giardia lamblia – cykl życiowy pasożyta
Przyczyną lambliozy (giardiozy) jest zarażenie pierwotniakiem Giardia lamblia(inaczej Giardia intestinalis, Giardia duodenalis). Jego rezerwuarem są zarówno domowe, jak i dzikie ssaki (np. psy, koty, bobry).
Cykl życiowy Giardia lamblia obejmuje dwa stadia rozwojowe: formę przetrwalną – cystę i formę wegetatywną – trofozoit. Poza organizmem pasożyty przeżywają wyłącznie w postaci cyst, które są odporne na niekorzystne warunki środowiskowe. Cysty mogą przetrwać w glebie lub zimnej wodzie do kilku miesięcy. Są też niewrażliwe na chlor stosowany do uzdatniania wody.
Cysty Giardia lamblia są dla człowieka formą inwazyjną – do zarażenia dochodzi drogą pokarmową poprzez ich połknięcie. W organizmie żywiciela cysty przemieszczają się z jamy ustnej do dwunastnicy, gdzie pod wpływem soku żołądkowego uwalniane są trofozoity zdolne do rozmnażania i uszkadzania jelita cienkiego. W kolejnym etapie cyklu trofozoity przesuwane do dalszych odcinków przewodu pokarmowego ponownie ulegają przekształceniu w cysty. W wydalanym kale obecne są zarówno cysty, jaki trofozoity.
Lamblioza (giardioza) – najczęstsze drogi rozprzestrzeniania zarażenia
Połknięcie skażonej cystami wody lub pokarmu
Bliski kontakt z osobą chorą na lambliozę, szczególnie w żłobkach, przedszkolach, domach dziecka
Podróżowanie po krajach o złych warunkach sanitarnych
Przenoszenie cyst do ust z zanieczyszczonych powierzchni (klamki w toalecie, pieluchy, przewijaki, zabawki)
Stosunki oralno-analne bez zabezpieczenia
Kontakt z zarażonymi zwierzętami lub środowiskiem zwierzęcym skażonym ich odchodami
Lamblioza (giardioza) – grupy ryzyka
Osoby w placówkach opieki nad dziećmi
Osoby z bliskiego kontaktu z chorym
Turyści w krajach o złych warunkach sanitarnych
Podróżnicy pijący nieoczyszczoną wodę ze źródeł, jezior lub rzek
Osoby zażywające kąpieli w zanieczyszczonych zbiornikach wodnych (baseny, rzeki, jeziora)
Ludzie czerpiący wodę do użytku domowego z płytkich studni
Osoby z osłabionym układem odpornościowym
Osoby mające kontakt z zarażonymi zwierzętami lub środowiskiem zwierzęcym skażonym ich odchodami
Lamblioza (giardioza) – rozwój choroby
Wystarczającą dawką prowadzącą do zarażenia jest połknięcie 10-100 cyst (w 1 g kału biegunkowego znajduje się 20 -150 tys. cyst). Chory/skolonizowany jest źródłem zarażenia dla osób z otoczenia. Wydalanie lamblii z kałem może trwać sześć miesięcy lub dłużej.
Najczęściej zarażenie Giardia lamblia prowadzi do bezobjawowej kolonizacji, która samoistnie ustępuje. Forma objawowa może przyjmować postać kliniczną ostrą lub przewlekłą.
Lamblioza (giardioza) ostra
Okres wylęgania ostrej lambliozy wynosi 7–14 dni. Objawy mogą utrzymywać się przez 2–4 tygodnie. Zazwyczaj choroba jest samoograniczająca się, ale u 30-50% nieleczonych zarażonych przechodzi w formę przewlekłą.
Objawy ostrej lambliozy:
biegunka (ponad 90% przypadków),
wzdęcia brzucha,
cuchnący, tłusty stolec bez krwi i śluzu,
·skurcze lub ból brzucha,
rozstrój żołądka lub nudności,
wymioty,
odwodnienie,
ubytek masy ciała.
Lamblioza (giardioza) przewlekła
Postać przewlekła może być następstwem ostrej postaci choroby lub pojawia się niezależnie od niej. Objawy są podobne jak w formie ostrej, ale symptomy są łagodniejsze i mają tendencję do ustępowania i powracania po kilku dniach lub tygodniach. Czasami u osób chorych na lambliozę występują długotrwałe powikłania, takie jak reaktywne zapalenie stawów, zespół jelita drażliwego i nawracająca biegunka, która może trwać latami. Ciężka lamblioza może prowadzić u dzieci do niedożywienia, opóźnienia rozwoju fizycznego i psychicznego, depresji.
Zachorowanie na lambliozę nie daje trwałej odporności, możliwe jest ponowne zarażenie i rozwój choroby.
Lamblioza (giardioza) – rozpoznanie
Diagnostyka lambliozy opiera się na kryteriach klinicznych, laboratoryjnych i epidemiologicznych. Zgodnie z definicją klasyfikacji przypadku, ustanowioną przez Komisję Wykonawczą Unii Europejskiej w 2018 r. potwierdzony przypadek lambliozy wymaga spełnienia kryteriów klinicznych i laboratoryjnych. W przypadku braku wyników badań laboratoryjnych, objawy kliniczne z powiązaniem epidemiologicznym wskazują jedynie na przypadek prawdopodobny.
Kryteria laboratoryjne:
Spełnienie co najmniej jednego z następujących trzech kryteriów:
wykazanie obecności cyst lub trofozoitówGiardia lamblia w stolcu, treści dwunastniczej lub w materiale z biopsji jelita cienkiego,
wykazanie obecności antygenuGiardia lamblia w stolcu, płynie dwunastniczym lub w materiale z biopsji jelita cienkiego,
wykrycie obecności kwasu nukleinowegoGiardia lamblia w stolcu, płynie dwunastniczym lub w materiale z biopsji jelita cienkiego.
Wykrycie w materiale od pacjenta cyst, trofozoidów, antygenu lub materiału genetycznego Giardia lamblia podlega zgłoszeniu do SANEPID-u zgodnie z ROZPORZĄDZENIEM MINISTRA ZDROWIA z dnia 24 czerwca 2020 r. w sprawie zgłaszania wyników badań w kierunku biologicznych czynników chorobotwórczych u ludzi.
Badanie laboratoryjne kału
Próbka:
Próbka kału jest najprostszym w pozyskaniu materiałem do badań laboratoryjnych w kierunku zarażeń Giardia lamblia. Do pobrania materiału nie potrzeba specjalnego przygotowania. Kał powinien być oddany do czystego naczynia (nocnik, nakładka na muszlę toaletową), skąd należy przenieść porcję wielkości orzecha włoskiego do naczynka kałowego (dostępne w Punktach Pobrań, aptekach). Próbkę należy jak najszybciej dostarczyć do laboratorium.
Ze względu na okresowe pojawianie się cyst w kale, w przypadku uzyskania wyniku ujemnego, badanie należy powtórzyć jeszcze dwukrotnie w odstępie 2-3 dni.
UWAGA: Potwierdzenie zarażenia u jednego z domowników jest wskazaniem do wykonania badania u pozostałych osób wspólnie zamieszkujących.
Lamblioza (giardioza) – leczenie
Leczenie lambliozy jest obowiązkowe u wszystkich zarażonych, zarówno przy wystąpieniu objawów, jak i w przypadku bezobjawowej kolonizacji. Terapia obejmuje leczenie objawowe biegunki i podawanie leków eliminujących pasożyta. Ze względu na możliwość nawrotu choroby, wskazane jest wykonanie badania kontrolnego (wykrywanie antygenu, mikroskopia) po upływie 2-4 tygodni od zakończenia leczenia.
Zapobieganie lambliozie (giardiozie)
Nie istnieją swoiste metody profilaktyki lambliozy.
Zapobieganiu zarażenia Giardia lamblia służy:
dbanie o higienę rąk,
czyszczenie i dezynfekcja powierzchni i przedmiotów,
unikanie spożycia wody bezpośrednio z jezior, rzek, strumieni,
spożywanie wody butelkowanej, gotowanej przez co najmniej 1 minutę lub filtrowanej przy braku pewności, co do bezpieczeństwa wody pitnej,
mycie owoców i warzyw w czystej wodzie,
unikanie spożywania pokarmów niewiadomego pochodzenia, szczególnie w krajach/miejscach o niskich standardach sanitarnych,
izolowanie osób z biegunką od pracy w placówkach opieki nad dziećmi,
DECYZJA WYKONAWCZA KOMISJI (UE) 2018/945 z dnia 22 czerwca 2018 r. w sprawie chorób zakaźnych i powiązanych szczególnych problemów zdrowotnych, które mają być objęte nadzorem epidemiologicznym, a także odpowiednich definicji przypadków – Dziennik Urzędowy Unii Europejskiej
ROZPORZĄDZENIEM MINISTRA ZDROWIA z dnia 24 czerwca 2020 r. w sprawie zgłaszania wyników badań w kierunku biologicznych czynników chorobotwórczych u ludzi.
Układ immunologiczny (odpornościowy) człowieka jest niezwykle skomplikowanym systemem, który odgrywa rolę „strażnika”, broniąc organizm przed obcymi patogenami, takimi jak wirusy i bakterie. Jednak czasami zdarza się, że ten sam system obronny, który nas chroni, może zacząć działać na niekorzyść organizmu i taki proces nazywany jest autoagresją. Polega ona na atakowaniu własnych komórek i tkanek, co prowadzi do rozwoju chorób autoimmunizacyjnych.
Czym są choroby autoimmunizacyjne?
Choroby autoimmunizacyjne są wynikiem zaburzeń w funkcjonowaniu układu immunologicznego, które powodują, że organizm rozpoznaje własne komórki i tkanki jako obce i wrogie, co prowadzi do ich zaatakowania. Przykłady takich chorób to toczeń rumieniowaty układowy (SLE, ang. Systemic Lupus Erythematosus), reumatoidalne zapalenie stawów, cukrzyca typu 1, autoimmunologiczne zapalenie wątroby i wiele innych. Objawy tych schorzeń mogą być różnorodne i początkowo zwykle dotyczą narządów, które w danym schorzeniu są atakowane, ale z czasem proces chorobowy zaczyna również obejmować inne organy wewnętrzne.
Choć dokładne przyczyny chorób autoimmunizacyjnych nie są w pełni poznane, to istnieje wiele teorii dotyczących ich rozwoju. Jedna z nich sugeruje, że niektóre osoby mogą wykazywać pewne predyspozycje genetyczne do rozwoju takich chorób, a czynniki środowiskowe, np. infekcje czy stres, przyczyniają się do ich aktywacji.
Badania i właściwe testy diagnostyczne
Rozpoznanie chorób autoimmunizacyjnych może być trudne, ponieważ ich objawy są często niespecyficzne i mogą przypominać inne schorzenia. Do potwierdzenia diagnozy konieczne jest zazwyczaj wykonanie szeregu badań laboratoryjnych, m.in. testów w kierunku autoprzeciwciał.
Autoprzeciwciała są istotnym elementem w zrozumieniu chorób autoimmunizacyjnych. To specyficzne białka wytwarzane przez układ immunologiczny, które atakują własne komórki i tkanki organizmu, zamiast skupiać się na zwalczaniu obcych patogenów. Badania autoprzeciwciał odgrywają kluczową rolę w wykrywaniu i monitorowaniu chorób autoimmunizacyjnych, często umożliwiając wczesną diagnozę, określenie stopnia aktywności choroby czy ocenę skuteczności leczenia.
Przeciwciała przeciwjądrowe ANA (ang. antinuclear antibodies) stanowią najszerszą i często badaną grupę autoprzeciwciał. Są one skierowane przeciwko antygenom zarówno jądra komórkowego, jak i cytoplazmy. Ich ocena jest kluczowym elementem diagnostyki chorób układowych tkanki łącznej.
Oficjalne rekomendacje jednoznacznie zalecają dwuetapową diagnostykę przeciwciał przeciwjądrowych ANA. Pierwszym etapem jesttest przesiewowy, który jest bardzo czuły. W tym przypadku zalecane są testy oparte o metodę immunofluorescencji pośredniej (IIFT, ang. indirect immunofluorescence technique), które są oceniane pod mikroskopem fluorescencyjnym. Doświadczony personel medyczny ocenia preparaty i w przypadku wykrycia przeciwciał przeciwjądrowych ANA rozpoznaje charakterystyczne typy świeceń, które odpowiadają poszczególnym autoprzeciwciałom. Na tym etapie można też określić poziom przeciwciał przeciwjądrowych ANA, czyli tzw. miano. Uzyskanie tej informacji jest niezwykle cenne z kilku względów:
poziom autoprzeciwciał często jest związany z aktywnością (zaawansowaniem) choroby
niskie miano przeciwciał przeciwjądrowych ANA (np. 1:100) może występować również u zdrowych osób
Wynik ujemny w teście IIFT jest ostateczny, podczas gdy wszelkie wyniki pozytywne wymagają potwierdzenia niezależną metodą.
Drugi etap diagnostyki, czyli etap potwierdzenia, opiera się na testach monospecyficznych, takich jak ELISA czy immunoblot, w których określa się specyficzność badanych przeciwciał. W przypadku diagnostyki przeciwciał przeciwjądrowych ANA istotne jest sprawdzenie, czy uzyskane wyniki są ze sobą spójne. Innymi słowy, przed podaniem ostatecznego wyniku, diagnosta laboratoryjny musi upewnić się, że wynik testu monospecyficznego jest zgodny z odpowiednim wzorcem fluorescencji uzyskanym na etapie badania przesiewowego.
Dla przykładu – przy podejrzeniu tocznia (SLE) zwykle wykonuje się badania w kierunku przeciwciał przeciwko:
Powyższe przeciwciała nie są wyłącznie przeciwciałami markerowymi, ale również mają znaczenie prognostyczne i mogą być stosowane do monitorowania przebiegu SLE (ich stężenie koreluje z aktywnością choroby).
Oprócz tego w kryteria rozpoznania tocznia rumieniowatego układowego (SLE) są włączone przeciwciała antyfosfolipidowe:
średnie lub wysokie miano przeciwciał antykardiolipinowych (IgA, IgG lub IgM),
dodatni test na obecność przeciwciał przeciwko ß2-glikoproteinie (IgA, IgG, IgM).
Podsumowanie
Choroby autoimmunizacyjne stanowią poważne wyzwanie zarówno dla lekarzy, jak i pacjentów. Ich zrozumienie wymaga dalszych badań naukowych nad mechanizmami układu immunologicznego oraz interakcji genetycznych i środowiskowych. Obecnie kluczową rolę odgrywają laboratoria medyczne, dostarczając precyzyjnych narzędzi do diagnozy i monitorowania tych schorzeń. Dla pacjentów ważne jest zdobycie wiedzy na temat swojej choroby, regularne kontrole oraz współpraca z zespołem medycznym w celu efektywnego zarządzania objawami i poprawy jakości życia.
Miedź (łac. cuprum) jest pierwiastkiem śladowym, czyli mikroelementem. Jej zasoby w organizmie człowieka określa się na 50-150 mg. Ciekawostką jest, iż łacińska nazwa miedzi pochodzi od wyspy Cypr, gdzie w czasach starożytnych wydobywano ten metal.
Pomimo faktu, iż zawartość miedzi w organizmie człowieka jest tak mała, jest ona elementem niezbędnym do prawidłowego funkcjonowania naszego ciała.
Wchłanianie miedzi w organizmie, transport miedzi we krwi i jej wydalanie
Proces wchłaniania miedzi odbywa się w jelicie cienkim. Aby było to możliwe, niezbędny jest proces poprzedzający, który zachodzi w żołądku i polega na uwolnieniu miedzi z połączeń organicznych.
Miedź po przejściu do dwunastnicy wchłaniania jest w enterocytach. Proces odbywa się w sposób aktywny, z udziałem białka nośnikowego CTR1. Wchłonięta miedź łączy się z albuminami osocza oraz transkupreiną. Następnie przenoszona jest do wątroby, gdzie z kolei łączy się z syntetyzowaną w tym narządzie ceruloplazminą, aby w postaci tego połączenia wrócić do krwiobiegu. Ceruloplazmina przenosi w osoczu ponad 90% miedzi, dlatego ma kluczowe znaczenie dla jej aktywności w organizmie.
Przyswajalność miedzi waha się od 5-50%, średnio wynosi 25-30%. Niedobór miedzi w organizmie zwiększa jej wchłanianie z diety, natomiast optymalny poziom miedzi w organizmie ogranicza to wchłanianie.
Miedź jest łatwiej przyswajana w obecności białka zwierzęcego oraz fruktozy. Utrudnione wchłanianie odbywa się w obecności pierwiastków, które konkurują z miedzią: żelazo, cynk, cyna, molibden, kadm, nikiel. Natomiast takie pierwiastki jak wapń czy fosfor utrudniają jej wydalanie.
Miedź słabiej się również wchłania przy alkalizacji treści żołądkowej. Dlatego zażywanie leków, które blokują wydzielanie kwasu solnego i działają alkalizująco – np. inhibitory pompy protonowej – utrudnia ten proces.
Miedź jest niezbędna do prawidłowego funkcjonowania komórek, a jej nadmiar jest toksyczny. Dlatego transport, dystrybucja i ilość biodostępnej miedzi musi być kontrolowana.
Dzienna podaż miedzi w diecie wynosi ok. 1,2 mg/dobę, co oznacza, iż wchłania się ok. 0,3-0,6 mg. Część miedzi, ok. 0,4 mg, wchłania się zwrotnie – z żółcią – do światła przewodu pokarmowego. Ok. 1 mg miedzi wydalany jest z kałem, bardzo niewielka część miedzi wydala się z moczem, potem i złuszczającym naskórkiem.
Rola miedzi w organizmie
Miedź jest składnikiem wielu enzymów, dzięki czemu wpływa na funkcjonowanie całego organizmu. Jest składnikiem m.in.:
ceruloplazminy – białka transportującego miedź, ale mającego również wpływ na homeostazę żelaza. CP uruchamia wątrobowe zapasy żelaza i wbudowywanie go w cząsteczkę transferyny;
dysmutazy ponadtlenkowej – enzymów SOD1 i SOD3. SOD1 jest enzymem zawierającym miedź i cynk, biorącym udział w procesach unieczynniania wolnych rodników i zmniejszającym stres oksydacyjny. SOD3 zwiększa ekspresję kolagenu, co w modelach na zwierzętach zwiększało m.in. przerost serca, poszerzanie lewej komory i zwłóknienie wywołane przeciążeniem wynikającym z nadciśnienia tętniczego;
oksydazy cytochromowej;
monoaminooksydazy – MAO – enzymu uczestniczącego w metabolizmie dopaminy do noradrenaliny, co ma wpływ na przesyłanie impulsów nerowych;
oksydazy lizynowej – enzymu uczestniczącego w odnowie białek i sieciowianiu kolagenu oraz lizyny.
Bezpieczne, dzienne spożycie miedzi wynosi do 3 mg, a okazjonalnie u osób dorosłych nawet 10 mg. Dawka toksyczna to ok. 20 g, a śmiertelna – 30 g.
Zawartość miedzi w diecie jest większa w produktach pochodzenia zwierzęcego niż roślinnego. W pierwszej grupie wynosi 0,05-0,35 mg/100 g, w drugiej 0,03-0,23 mg/100g. Z produktów pochodzenia zwierzęcego najwięcej miedzi zawiera wątróbka. Pokarmy roślinne to ziarna słonecznika, nasiona soi, orzechy laskowe, płatki owsiane, rośliny strączkowe (groch, fasola).
Należy pamiętać, że w miejscach, gdzie jest miedziana instalacja dostarczająca wodę, dostarczanie miedzi jest większe. Więcej tego pierwiastka znajduje się w wodzie ciepłej niż zimnej.
Miedź powinna być dostarczana z pożywienia. Suplementacja miedzi w stanach, gdy nie ma zwiększonego zapotrzebowania może być toksyczna. Należy również pamiętać, że miedź i inne mikroelementy – zwłaszcza cynk – muszą być dostarczane w równowadze. Zwiększenie ilości miedzi w diecie to obniżenie wchłaniania cynku i odwrotnie.
Liu Y, Miao J. An Emerging Role of Defective Copper Metabolism in Heart Disease. Nutrients. 2022 Feb 7;14(3):700. doi: 10.3390/nu14030700. PMID: 35277059; PMCID: PMC8838622.
Gertig H., Przysławski J., Bromatologia. Zarys nauki o żywności i żywieniu. Wydawnictwo Lekarskie PZWL, Warszawa 2015.


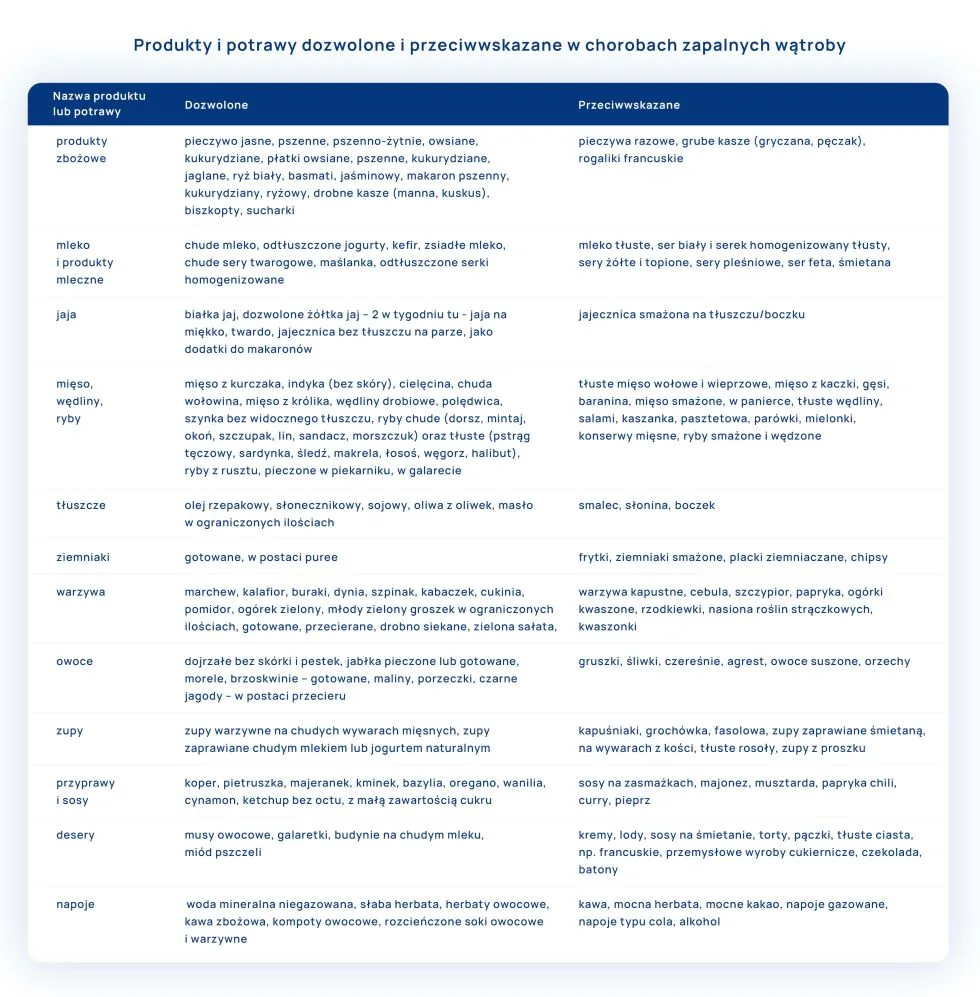

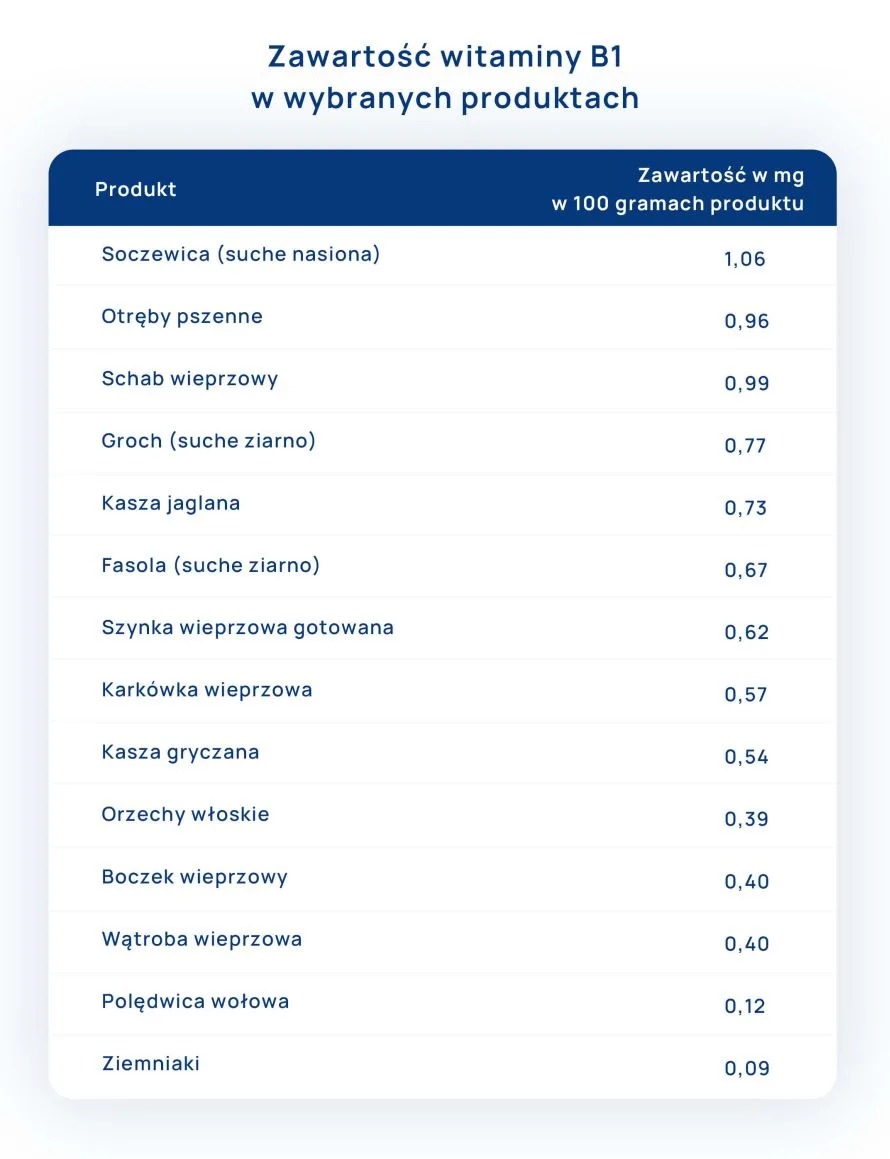
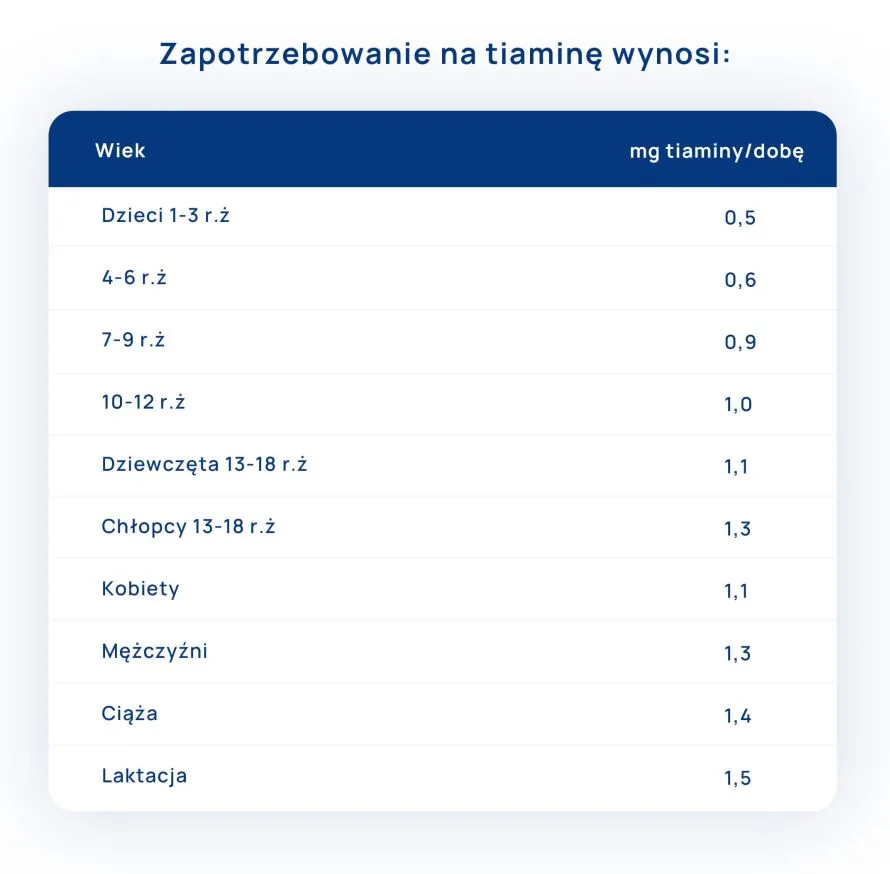










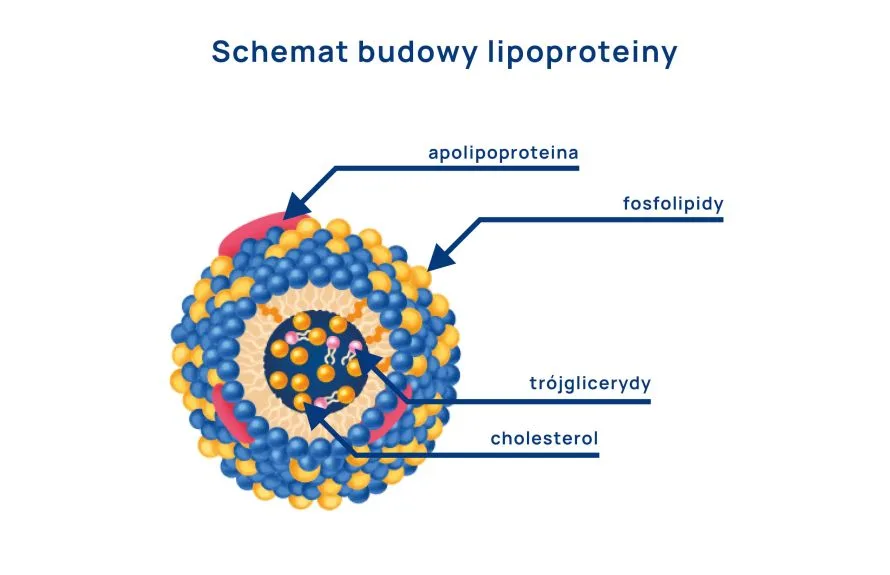
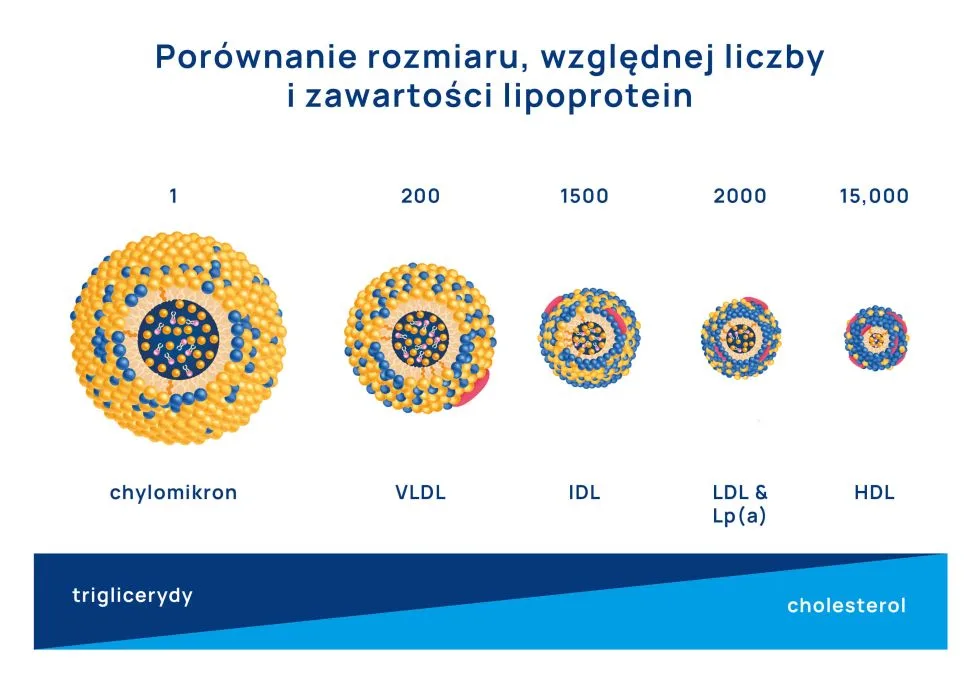
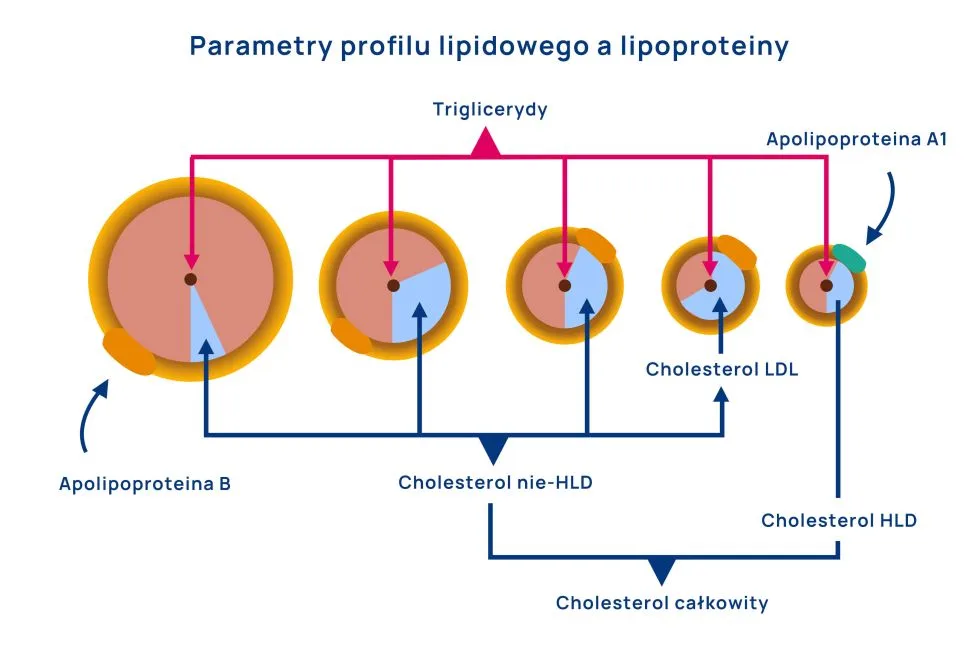
 – dawniej niealkoholowe stłuszczenie wątroby (NAFLD)")
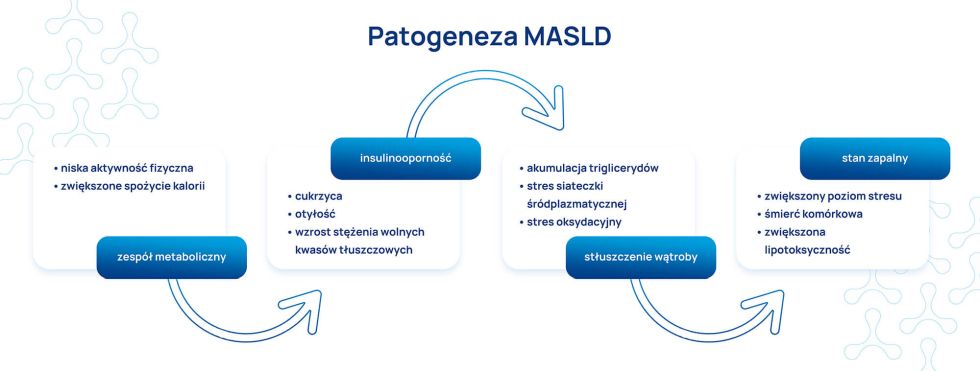
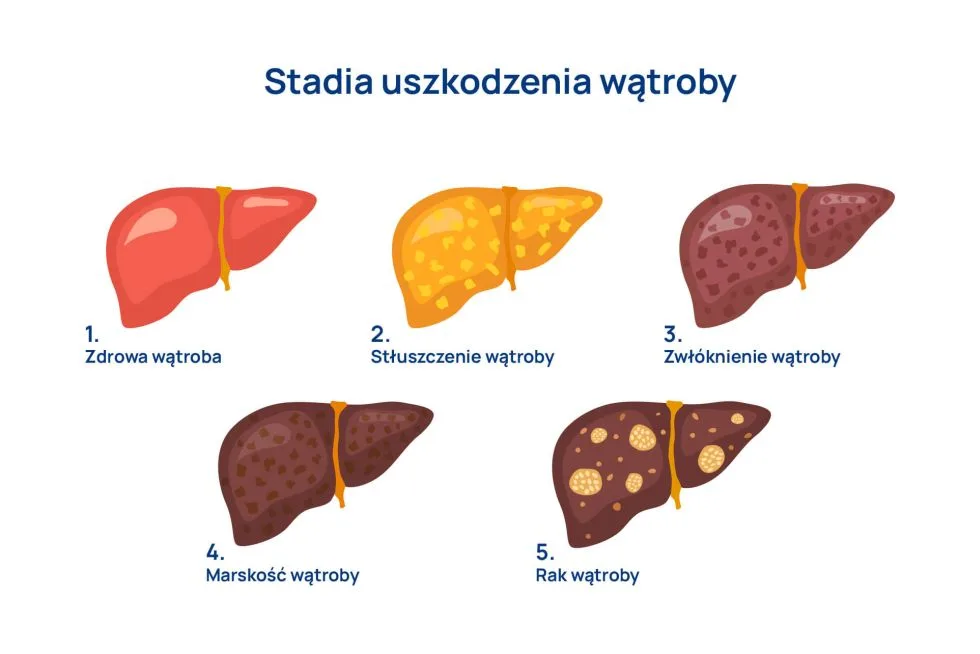

 – warto wiedzieć więcej")