Inkretyny rewolucjonizują podejście do leczenia cukrzycy. Artykuł przybliża, czym są hormony inkretynowe, czym jest efekt inkretynowy, jak inkretyny działają w cukrzycy i czy wykazują działania pozatrzustkowe.
Spis treści
- Inkretyny – czym są? Mechanizm działania inkretyn
- Hormony inkretynowe – wydzielanie
- Funkcje hormonów inkretynowych
- Leki na cukrzycę nowej generacji
- Leki inkretynowe
Inkretyny – czym są? Mechanizm działania inkretyn
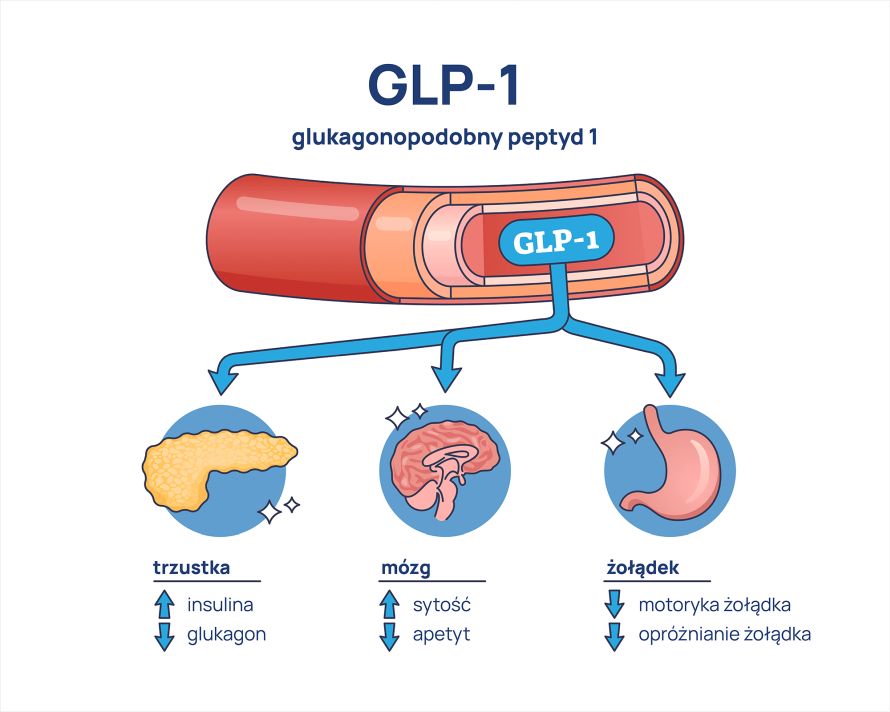
Hormony inkretynowe, zwane też inkretynami, to hormony produkowane przez komórki błony śluzowej jelit. Mechanizm ich działania polega na tym, iż po spożyciu pokarmu pobudzają trzustkę (komórki β trzustki) do uwalniania insuliny, dzięki czemu obniża się poziom cukru we krwi. Ponadto blokują wydzielanie glukagonu, hamują opróżnianie żołądka i ograniczają łaknienie oraz pobieranie pokarmu.
Głównymi hormonami inkretynowymi są:
- glukagonopodobny peptyd 1 (GLP-1)
- glukozozależny polipeptyd insulinotropowy (GIP, ang. glucose-dependent insulinotropic peptide).
GLP-1 i GIP są hormonami, które działają w sposób współzależny i wzmacniają wzajemnie swoje efekty.
U zdrowych osób uwalnianie GLP-1 w odpowiedzi na posiłek następuje bardzo szybko (poniżej 10 minut) i jest skorelowane z wydzielaniem insuliny do układu krążenia wrotnego. U osób chorujących na cukrzycę typu 2, lub wykazujących nieprawidłową tolerancję glukozy, odpowiedź ta jest nieprawidłowa i prowadzi do obniżenia poposiłkowego stężenia GLP-1 oraz zmniejszonego wydzielania insuliny po jedzeniu.
Efekt działania tych hormonów na metabolizm glukozy – tzw. efekt inkretynowy – jest kluczowy w regulacji gospodarki węglowodanowej u osób zdrowych, odpowiada za ok. 50-70% insulinowej reakcji na posiłek.
| WAŻNE Hormony inkretynowe stymulują wydzielanie insuliny, zapobiegając zbyt dużemu wzrostowi glikemii poposiłkowej. Ponadto spowalniają opróżnianie żołądka i przyczyniają się do zmniejszenia apetytu, co również pomaga w kontrolowaniu poziomu cukru we krwi. |

Hormony inkretynowe – wydzielanie
Oba hormony inkretynowe (inkretyny) są wytwarzane i wydzielane w jelicie cienkim. Stężenie GLP-1 i GIP wzrasta gwałtownie podczas posiłku. Ich okres półtrwania jest bardzo krótki, nie przekracza 2 minut, dlatego są bardzo szybko usuwane z krążenia, w czym uczestniczy enzym dipeptydylopeptydazę IV (DPP-4). Tkanką docelową obydwu hormonów jest trzustka, jednak ich receptory można znaleźć również poza nią.
- Receptory GIP: znajdują się przede wszystkim na komórkach beta trzustki, ale także w tkance tłuszczowej i ośrodkowym układzie nerwowym.
- Receptory GLP-1: są obecne na komórkach alfa i beta wysp trzustkowych, w ośrodkowym i obwodowym układzie nerwowym, sercu, płucach, przewodzie pokarmowym oraz nerkach.
Insulinotropowe działanie GLP-1 jest zależne od poziomu glukozy – wymaga stężeń glukozy przekraczających 90 mg/dl (5 mmol/l), aby nasilić wydzielanie insuliny i zahamować wydzielanie glukagonu. Ważne jest, iż organizm zachowuje pełną zdolność do uwalniania glukagonu w odpowiedzi na hipoglikemię, nawet w obecności GLP-1.
Enzym dipeptydylopeptydaza IV (DPP-4) rozkłada GLP-1 do amidu GLP-1, co całkowicie inaktywuje jego aktywność w stosunku do stymulacji wydzielania insuliny. Jednak wspomniany amid GLP-1 może mieć korzystne działanie kardiologiczne, w którym pośredniczą mechanizmy niezwiązane z receptorem GLP-1.
Funkcje hormonów inkretynowych
Kluczowe znaczenie inkretyn to ich działania insulinotropowe, czyli udział w kontroli glikemii, nie jest to jednak jedyny wpływ na funkcje organizmu.
Wpływ na apetyt
GLP-1 odgrywa kluczową rolę w regulacji apetytu i sytości. Jego działanie obejmuje:
- zmniejszenie łaknienia – ogranicza chęć do jedzenia,
- zwiększenie uczucia sytości – przedłuża czas, w którym pacjent czuje się najedzony,
- spowolnienie opróżniania żołądka – dzięki temu pokarm dłużej pozostaje w żołądku, co dodatkowo wzmacnia uczucie sytości.
W przeciwieństwie do GLP-1, GIP nie wydaje się mieć istotnego wpływu na odczuwanie głodu i sytości.
>>> Przeczytaj też: Leptyna – hormon głodu i sytości
Wpływ na masę ciała
GLP-1 ma również korzystny wpływ na metabolizm tłuszczów – efekt inkretynowy u osób otyłych jest zmniejszony, nawet jeśli nie występują zaburzenia tolerancji glukozy. Badania wskazują, że podawanie agonistów receptora GLP-1 (GLP-1RA) może zmniejszać grubość białej tkanki tłuszczowej, a także redukować zawartość niebezpiecznej tkanki tłuszczowej trzewnej. Istnieją także obserwacje sugerujące jego wpływ na „brązowienie” białej tkanki tłuszczowej, co mogłoby zwiększać wydatek energetyczny organizmu, choć potrzebne są dalsze badania w tym zakresie.
Rola GIP w kontekście masy ciała jest nadal badana. Wyniki z modeli zwierzęcych sugerują, że myszy pozbawione receptora GIP (GIPR) nie rozwijają otyłości, nawet przy diecie wysokotłuszczowej, co wskazuje na związek GIP z gromadzeniem tkanki tłuszczowej.

>>> Przeczytaj też: Otyłość i jej zapobieganie – profilaktyka otyłości pierwotnej i wtórnej
Leki na cukrzycę nowej generacji
Hormony inkretynowe (GIP i GLP-1) odgrywają ważną rolę w kontoli glikemii, w cukrzycy typu 2 poprzez przeciwdziałanie na:
- zmniejszony lub całkowicie wyłączony efekt inkretynowy, który jest jednym z czynników patofizjologicznych tego procesu,
- pogorszenie poposiłkowej kontroli glikemii w wyniku, która wpływa na progresję i rozwój choroby.
Potencjał terapeutyczny GLP-1 został z powodzeniem wykorzystany w opracowaniu agonistów receptora GLP-1. Efektem tych badań było opracowanie leków hipoglikemizujących opartych na:
- selektywnych agonistach receptorów GLP-1 – substancjach wybiórczo stymulujących tylko receptory GLP-1, należy do nich semaglutyd. Ich działanie polega na naśladowaniu działania naturalnego GLP-1,
- koagonistach – substancjach stymulujących zarówno receptory GLP-1 jak GIP, należy do nich tirzepatyd. Wykazują jeszcze skuteczniejsze działanie w redukcji poziomu HbA1c (wskaźnika długoterminowej kontroli cukrzycy) oraz masy ciała w porównaniu do selektywnych agonistów GLP-1.
| CIEKAWOSTKA Na działanie inkretyn znacząco wpływają operacje bariatryczne, np. wyłączenie żołądkowo-jelitowe na pętli Roux-en-Y (RYGB). Jest to zabieg chirurgiczny, w którym część żołądka i dwunastnicy zostaje ominięta, a jelito cienkie zostaje przyłączone do małego zbiornika żołądka, tworząc kształt litery „Y”. Po RYGB obserwuje się wzrost stężenia GLP-1, co przyczynia się do poprawy kontroli glikemii i przywrócenia efektu inkretynowego u pacjentów z cukrzycą typu 2. |

>>> Przeczytaj też: Nadwaga a otyłość – czym się różnią? Klasyfikacja, leczenie i konsekwencje otyłości
Leki inkretynowe
Leki inkretynowe zarejestrowane w leczeniu cukrzycy dzielą się na:
- GLP-1RA – analogi receptora peptydu glukagonopodobnego 1 (GLP-1) – zwiększają kontrolę gkliemii oraz zmniejszają masę ciała,
- inhibitory DPP-4 – kontrolują glikemię, nie mają wpływu na masę ciała.
Do grupy leków GLP-1 należą:
- semaglutyd – obecny w preparacie Ozempic – podawany raz w tygodniu, w postaci iniekcji, zarejestrowany w leczeniu cukrzycy typu 2, oraz w leku Rybelsus (dostępny w postaci tabletek). Semaglutyd jako substancja aktywna może być również stosowany w leczeniu otyłości,
- liraglutyd – obecny w preparacie Saxenda – podawany raz w tygodniu w postaci iniekcji, zarejestrowany w leczeniu otyłości,
- liksysenatyd – obecny w preparacie Suliqua, który zawiera również insulinę, podawany we wstrzyknięciach,
- duaglutyd,
- eksenatyd.
Inhibitory DPP-4 to:
- sitagliptina,
- saksagliptyna,
- linagliptyna,
- alogliptyna,
- wildagliptyna.
Podsumowanie
Wiedza o hormonach inkretynowych oraz wykorzystanie ich potencjału terapeutycznego zrewolucjonizowało leczenie cukrzycy. Niestety efekt w postaci obniżania masy ciała, spowodował, iż leki GLP-1 są nadużywane, co może prowadzić do poważnych problemów zdrowotnych. Długoterminowe konsekwencje niekontrolowanego stosowania tych preparatów nie są jeszcze w pełni poznane.
>>> Przeczytaj też: Otyłość brzuszna (trzewna) u kobiet i mężczyzn – przyczyny, objawy, diagnostyka i leczenie
Piśmiennictwo
- Michałowska J., Bogdański P., Rola hormonów i leków inkretynowych w terapii otyłości i wybranych zaburzeń, Forum Zaburzeń Metabolicznych 2021, vol. 12, no 2, 61–69
- M.A. Nauck, T.D. Müller: Incretin hormones and type 2 diabetes, Diabetologia 2023,66.(Review)






: objawy, przyczyny, diagnostyka i leczenie")

: przyczyny, objawy i leczenie zespołu wstrząsu toksycznego")

: objawy, przyczyny, diagnostyka i leczenie")